大家好,今天为大家介绍的是仓鼠在揭示朊病毒致痴呆中的贡献。
斯坦利·普鲁西纳因揭示疯牛病、羊瘙痒病、克-雅二氏病和类克-雅二氏病等神经退行性疾病的致病机制而闻名。他首次提出并验证朊病毒(Prion)作为不含核酸的蛋白质感染颗粒,开创性地更新了人类对病原体类型的认知,这一发现为相关疾病的诊断与治疗提供了科学基础。并获得1997年诺贝尔生理学或医学奖。
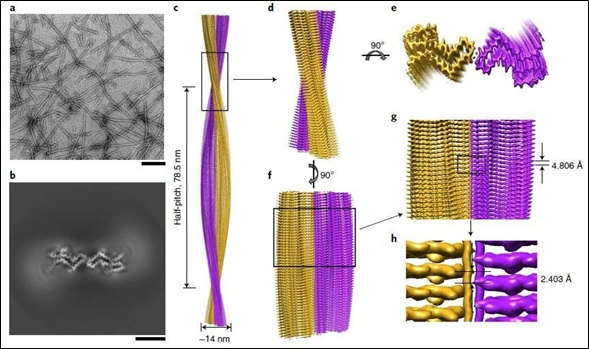
全长人朊病毒蛋白纤维的冷冻电镜结构[1]
1. 朊病毒导致痴呆(主要指克雅病)的核心作用机制
(1)病原体:朊蛋白(Prion Protein)的形成
正常形式(PrP^C)是一种存在于所有哺乳动物神经元细胞膜表面的正常蛋白质,功能尚未完全明确,可能与神经保护、突触功能有关。它具有可溶性,可被蛋白酶降解。异常形式(PrP^Sc)是PrP^C的错误折叠异构体。其空间构象发生改变,富含β-折叠,具有抗蛋白酶水解的特性,且不溶于变性剂。
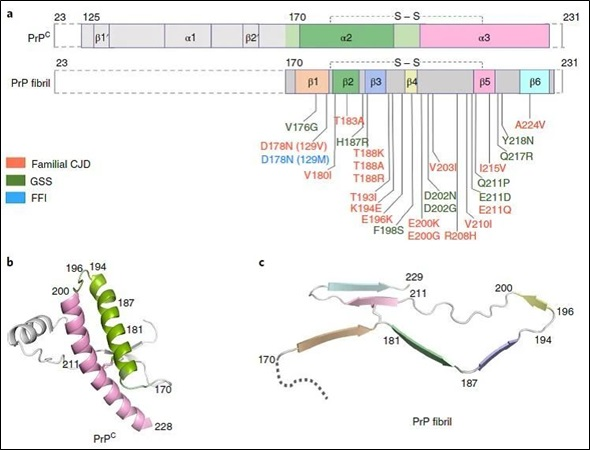
人野生型PrPC和PrP纤维结构比较[1]
(2)“传染”与增殖(核-聚合模型)
①模板诱导:外源性或内源性产生的PrP^Sc作为“种子”或模板,与相邻的正常PrP^C分子接触。
②构象转化:PrP^Sc通过物理相互作用,强行改变PrP^C的构象,使其“复制”成与自己一样的错误折叠结构(即新的PrP^Sc)。
③链式反应与聚合:新产生的PrP^Sc分子又可以作为模板去转化更多的PrP^C,形成一个指数级增长的链式反应。这些新生成的PrP^Sc分子会相互聚集。
(3)神经毒性产生
①聚集与沉积:大量产生的PrP^Sc分子聚集形成不溶性的寡聚体和原纤维,最终在神经元内和细胞间形成淀粉样斑块沉积。
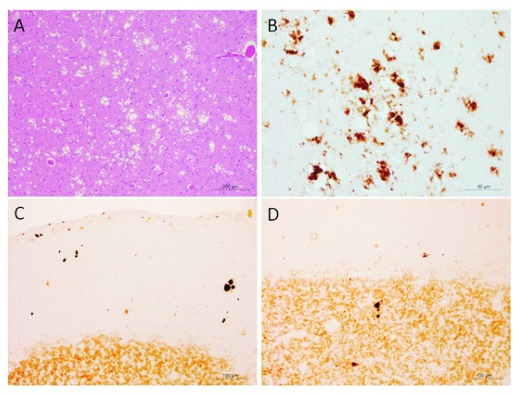
异常朊蛋白沉积[2]
②直接与间接损伤:
破坏细胞功能:异常聚集的PrP^Sc可能直接干扰神经元的离子通道、突触传递、蛋白质稳态等关键功能;
激活病理通路:触发内质网应激、线粒体功能障碍、氧化应激和炎症反应等;
胶质细胞活化:持续的聚集和细胞损伤会激活大脑中的免疫细胞(小胶质细胞),释放炎症因子,加剧神经损伤。
③神经元死亡与空泡变性:最终导致神经元大量凋亡或坏死。死亡神经元被清除后,留下微小的空泡,使脑组织在显微镜下呈“海绵状”(海绵样变)。
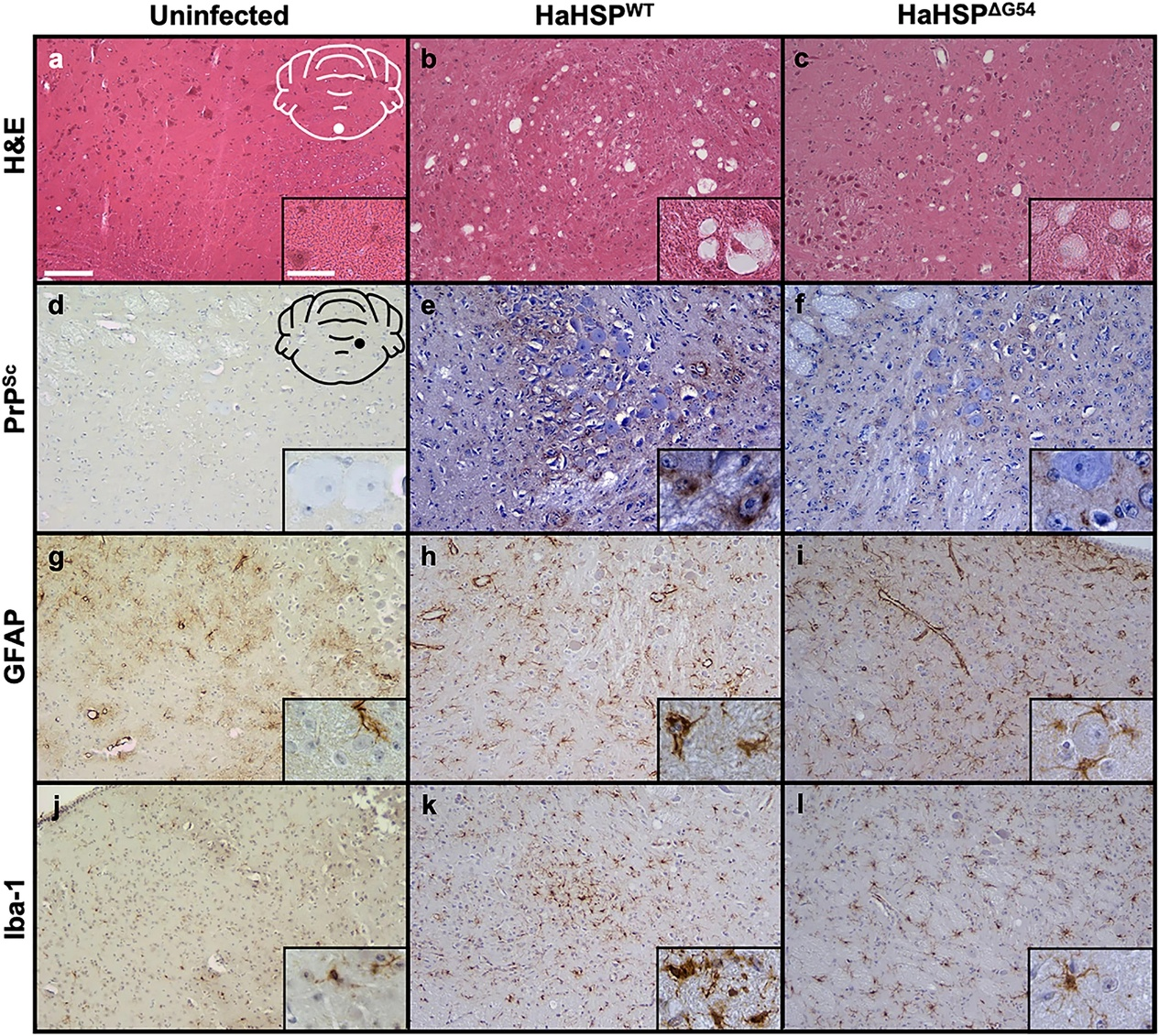
海绵样变[3]
(4)临床表现
由于上述过程广泛而不可逆地损害大脑皮层、基底节、丘脑等与认知、记忆、运动、情感相关的脑区,患者会迅速出现进行性加重的痴呆、肌阵挛、共济失调、视觉障碍,最终陷入昏迷,直至死亡。
2. 仓鼠在揭示朊病毒致痴呆方面的突出贡献

(1)确立“株系”概念,证明朊病毒是信息载体
在20世纪80年代,科学家将不同来源的朊病毒(如来自仓鼠、小鼠)进行交叉接种实验。
关键发现:仓鼠适应株朊病毒接种到仓鼠体内后,表现出高度稳定且可重复的疾病特征:潜伏期极短(约70天,远短于小鼠的150天以上)、大脑中朊病毒滴度极高、且神经病理分布模式固定。
(2)提供了高效的生物检测系统
由于仓鼠感染后潜伏期短、脑中病毒滴度极高,使得从脑组织中提取和纯化朊病毒变得非常高效。这为后续生化分析(如发现PrP^Sc对蛋白酶K有抗性)、结构研究以及检测方法开发(如免疫印迹)提供了大量、可靠的实验材料,是实验室研究的“生产工厂”。
(3)揭示了物种屏障与朊病毒复制的机制
通过研究仓鼠与小鼠之间的交叉感染,科学家明确了“物种屏障”的存在:跨物种传播效率低、潜伏期长。
进一步研究发现,屏障强度与宿主正常朊蛋白(PrP^C)与入侵的PrP^Sc之间氨基酸序列的相似度高度相关。仓鼠模型为理解朊病毒如何适应新宿主、以及复制所需的分子匹配机制提供了直观证据。
(4)作为治疗和消毒效果测试的平台
仓鼠模型因其快速的病程,成为筛选抗朊病毒药物、评估消毒剂和手术器械灭菌方法有效性的理想活体测试系统。
3. 朊病毒对人类产生的巨大意义
(1)突破了“中心法则”,朊病毒的发现证明,蛋白质本身可以作为遗传信息的载体;重新定义“传染源”:首次证明一种不含核酸的纯蛋白质颗粒就具有传染性。
(2)为理解人类常见神经退行性疾病开辟了新范式,提供了“蛋白质错误折叠病”的统一机制模型,揭示了“病理蛋白在脑内类似朊病毒的播散”机制 。
(3)对公共卫生和医学安全的深远影响,革新了医疗器械消毒和外科手术规范,保障了血液和生物制品安全
(4)对食品安全和畜牧业管理的重大警示,直接催生了针对“疯牛病(BSE)的全球性防控
(5)推动前沿科学和技术的发展,促进新的检测技术并启发新的治疗思路
今天的科普到这里就结束了,我们下期再见。
4. 参考文献
[1] WANG L Q, ZHAO K, YUAN H Y, et al. Cryo-EM structure of an amyloid fibril formed by full-length human prion protein [J]. Nat Struct Mol Biol, 2020, 27(6): 598-602.
[2] YAARI R, FAVARO S, BURKE A D, et al. A rapid turn of events: sudden physical and cognitive decline [J]. Prim Care Companion CNS Disord, 2013, 15(3).
[3] BLOCK A J, YORK T C, BENEDICT R, et al. Prion protein amino acid sequence influences formation of authentic synthetic PrP(Sc) [J]. Sci Rep, 2023, 13(1): 441.
[4] Bolton, D. C., McKinley, M. P., & Prusiner, S. B. (1982). Identification of a protein that purifies with the scrapie prion. Science, *218*(4579), 1309-1311.
[5] Prusiner, S. B., et al. (1987). Distinct prion proteins in short and long scrapie incubation period mice. Cell, *51*(4), 651-662.
[6] Meyer, R. K., et al. (1986). Separation and properties of cellular and scrapie prion proteins. Proceedings of the National Academy of Sciences, *83*(8), 2310-2314.
[7] Scott, M., et al. (1993). Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell, *73*(5), 979-988.
[8] Race, R., et al. (1995). Temporal and spatial control of PrP replication in transgenic mice. Neuron, *14*(4), 771-781.
[9] Kong, Q., et al. (2004). Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathologica, *107*(3), 273-280.
[10] Kirby, L., et al. (2011). Persistence of pathogenic prion protein during simulated wastewater treatment processes. Environmental Science & Technology, *45*(19), 10125-10132.
[11] Nichols, T. A., et al. (2009). Resistance of soil-bound prions to rumen digestion. PLoS ONE, *4*(8), e6738.
[12] White, M. D., et al. (2008). RNAi therapy for prion disease in a mouse model. Journal of Clinical Investigation, *118*(8), 2682-2690.
[13] Peretz, D., et al. (2006). Inactivation of prions by acidic sodium dodecyl sulfate. Journal of Virology, *80*(1), 322-331.

