一个文献的cytof数据集,标题是:《Single‑cell profiling of myasthenia gravis identifies a pathogenic T cell signature》,这个文献的cytof数据在:https://data.mendeley.com/datasets/nkcb8nc7w8/1,感兴趣的也可以自行下载进行处理。
队列是:peripheral blood mononuclear cells (PBMCs) from myasthenia gravis patients (MG, n = 38) and healthy controls (CTRL, n = 21)
• Thymic leukocytes from MG patients (n = 4) and non-MG incidental mass lesion controls (n = 6)
• Thymic tissue sections of MG patients (n = 13) and non-MG controls (n = 6)
有两种CyTOF:
• surface markers
• intracellular cytokines (following brief antigen-independent restimulation)
发现它居然就是单独的fcs文件,如下所示:
$ ls -lh ../dataFiles/ |cut -d" " -f 5-2.4K Mar 24 2021 CyTOF_blood_live_ICS_metadata.csv1.1G Mar 24 2021 CyTOF_blood_live_ICS_untrans_merged.fcs2.3K Mar 24 2021 CyTOF_blood_live_surf_metadata.csv966M Mar 24 2021 CyTOF_blood_live_surf_untrans_merged.fcs
也就是说,这个文献里面的两个队列,多个病人样品的cytof数据,被合并为同一个文件啦。确实有点麻烦,我使用下面的代码进行了简单的探索:
require(cytofWorkflow)c1 = read.flowSet('../dataFiles/CyTOF_blood_live_ICS_untrans_merged.fcs')# A flowSet with 1 experiments.c1 # flowFrame object c1[[1]]c1=c1[[1]]# expression valuesexprs( c1 )[1:6, 1:5]dim(exprs( c1 ))as.character(colnames(exprs(c1)))
主要是flowFrame这个对象的理解,对象都是复杂的,但是这个flowFrame对象最重要的其实就是矩阵,里面是700万个单细胞的30多个抗体的信号值矩阵,所以我使用了下面的代码进行拆开:
exp_list = split(as.data.frame( exprs( c1 ) ), exprs( c1 )[,37])names(exp_list) = paste0('p',names(exp_list))names(exp_list)dir.create('new') lapply(names(exp_list) , function(x){ # x = names(exp_list)[[1]];x tmp = c1 tmp@exprs = as.matrix(exp_list[[x]] ) write.FCS(tmp,file.path('new',paste0(x,'.fcs')))})
把每个样品都输出自己的fcs文件,输出如下所示的文件:
$ ls -lh new/|cut -d" " -f 5- 20M Feb 7 16:52 p1.fcs 20M Feb 7 16:52 p10.fcs 20M Feb 7 16:52 p11.fcs5.0M Feb 7 16:52 p12.fcs 21M Feb 7 16:52 p13.fcs 22M Feb 7 16:52 p14.fcs 21M Feb 7 16:52 p15.fcs 16M Feb 7 16:52 p16.fcs 18M Feb 7 16:52 p17.fcs 15M Feb 7 16:52 p18.fcs 14M Feb 7 16:52 p19.fcs
每个fcs后缀的文件,都是单独一个样品的cytof数据文件,里面都是十多万个单细胞哦!
然后仍然是批量读取:
p1='new'fs1=list.files(p1,'*fcs' )fs1samp <- read.flowSet(files = fs1,path = p1)
读取后就可以进行我们前面的教程处理啦,教程链接合辑是:
• 1.cytof数据资源介绍(文末有交流群)
• 2.cytofWorkflow之读入FCS文件(一)
• 3.cytofWorkflow之构建SingleCellExperiment对象(二)
• 4.cytofWorkflow之基本质量控制(三)
• 5.cytofWorkflow之聚类分群(四)
• 6.cytofWorkflow之人工注释生物学亚群(五)
• 7.cytofWorkflow之亚群比例差异分析(六)
可以看到绝大部分样品都是细胞数量在10万附近:
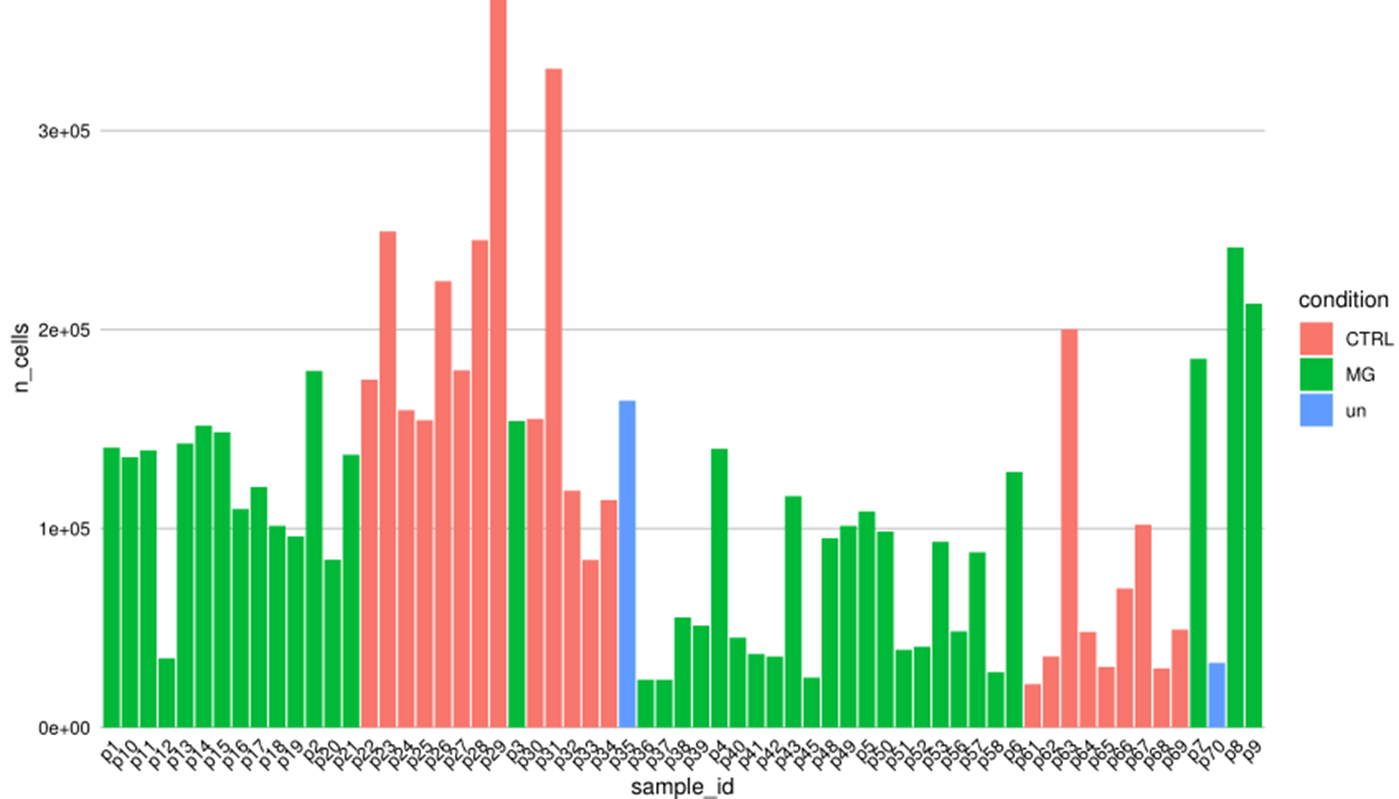
细胞数量在10万
而且绝大部分都是T细胞,包括CD4和CD8的T细胞:
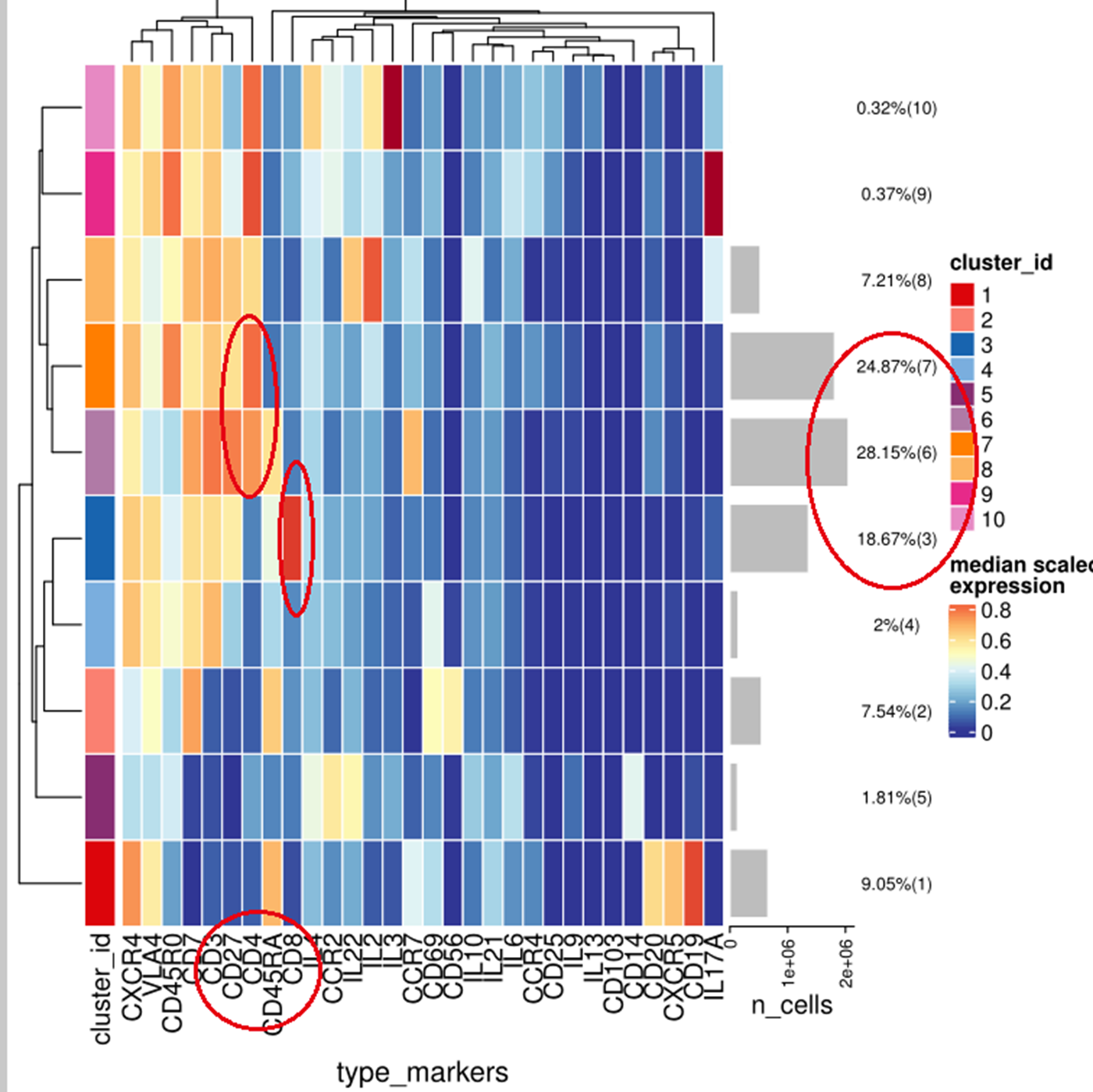
绝大部分都是T细胞
但是如果要做到文章那样的降维聚类分群和生物学命名,还是有点难度哦:
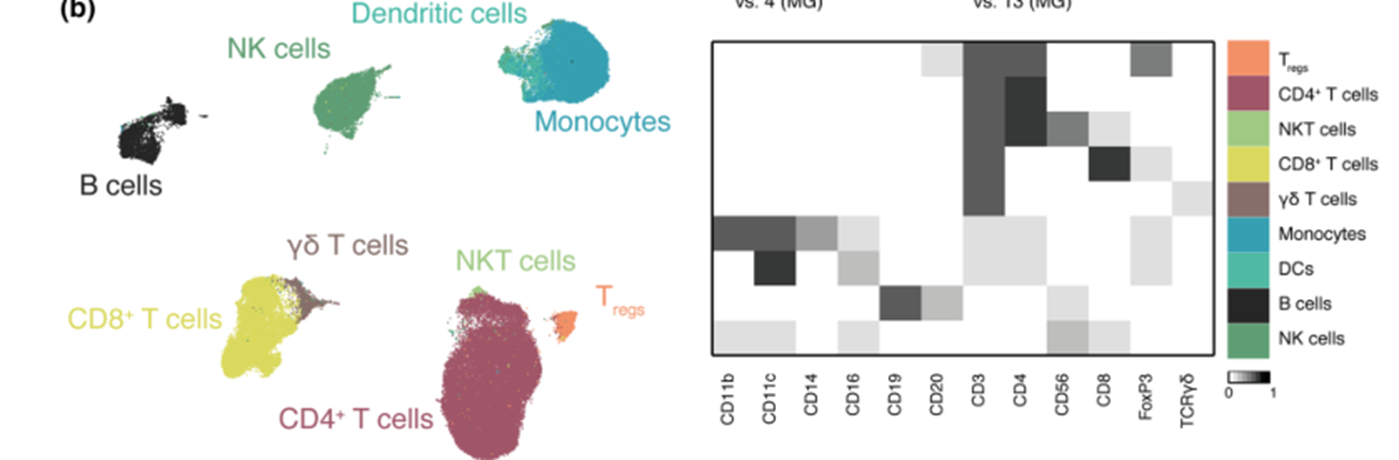
文章那样的降维聚类分群和生物学命名
转载自《生信技能树》,如有侵权,请联系删除。

