PRJNA764023 数据集的 fastq 原始数据,到新格元的官方网站,有对这款试剂盒及其分析软件(celescope)的介绍,在 github 上有软件的使用说明及下载:https://github.com/singleron-RD/CeleScope
其官方文档也很清晰:https://github.com/singleron-RD/CeleScope/blob/master/docs/quick_start.md
在新格元的 GitHub 里面下载 CeleScope
我们这里直接使用 conda 安装即可;你的服务器空白用户里面安装自己的 conda,每个用户独立操作,安装方法代码如下:
# 首先下载文件,20M/S的话需要几秒钟即可wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh# 接下来使用bash命令来运行我们下载的文件,记得是一路yes下去bash Miniconda3-latest-Linux-x86_64.sh # 安装成功后需要更新系统环境变量文件source ~/.bashrc
接下来 使用 conda 安装 aspera,后面我们下载 PRJNA764023 数据集的 fastq 文件用得着,代码如下:
conda create -n download conda activate download conda install -y -c hcc aspera-cliconda install -y -c bioconda sra-toolswhich ascp ## 一定要搞清楚你的软件被conda安装在哪ls -lh ~/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh
我们已经多次介绍过 conda 细节了,这里就不再赘述。
• conda 管理生信软件一文就够
• 生信技能树 B 站软件安装视频
• https://www.bilibili.com/video/av28836717
但是新格元的 CeleScope 软件需要重新建立一个 conda 环境。
git clone https://github.com/singleron-RD/CeleScope.git cd CeleScope# 其GitHub提供了一个 conda_pkgs.txt 文件,所以需要首先 git clone 它conda install mambamamba create -n celescope -y --file conda_pkgs.txt####这个txt文件中包含了运行这个软件所需要的其他软件的版本信息 conda activate celescopepip install celescope
也就是说,本来是应该是很简单的 pip install 安装一个 Python 模块,结果因为它太多的依赖,我们怕 pip install 失败,所以首先使用 conda 新建了一个 celescope 的小环境,在这个小环境里面安装了大量的依赖,最后才小心翼翼的 pip install celescope
测试 CeleScope 软件
官方教程提供了一个小数据来测试这款软件是否安装好了:https://github.com/singleron-RD/celescope_test_script
mkdir test_dircd test_dirgit clone https://github.com/singleron-RD/celescope_test_data.gitgit clone https://github.com/singleron-RD/celescope_test_script.gitpip install pytestcd celescope_test_scriptpython -m pytest -s test_multi.py --assays rna####我们要跑的是rna数据,所以--assays后面写rnacd celescope_test_scriptpython -m pytest -s test_multi.py
如果测试成功了,返回 success 的结果。
正式使用 CeleScope 软件
首先需要下载参考基因组数据和基因注释文件
我们的物种是人类,这里就很简单了:
mkdir hs_ensembl_99cd hs_ensembl_99wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gzwget ftp://ftp.ensembl.org/pub/release-99/gtf/homo_sapiens/Homo_sapiens.GRCh38.99.gtf.gzgunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gzgunzip Homo_sapiens.GRCh38.99.gtf.gzconda activate celescopecelescope rna mkref \ --genome_name Homo_sapiens_ensembl_99 \ --fasta Homo_sapiens.GRCh38.dna.primary_assembly.fa \ --gtf Homo_sapiens.GRCh38.99.gtf
得到的参考基因组后续用得上
接下来需要下载 PRJNA764023 数据集的 fastq 文件
制作如下所示 文件名 fq.txt 是的文本文件,内容如下所示:
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/076/SRR15927876/SRR15927876_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/076/SRR15927876/SRR15927876_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/077/SRR15927877/SRR15927877_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/077/SRR15927877/SRR15927877_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/078/SRR15927878/SRR15927878_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/078/SRR15927878/SRR15927878_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/079/SRR15927879/SRR15927879_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/079/SRR15927879/SRR15927879_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/080/SRR15927880/SRR15927880_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/080/SRR15927880/SRR15927880_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/081/SRR15927881/SRR15927881_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/081/SRR15927881/SRR15927881_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/082/SRR15927882/SRR15927882_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/082/SRR15927882/SRR15927882_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/083/SRR15927883/SRR15927883_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/083/SRR15927883/SRR15927883_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/084/SRR15927884/SRR15927884_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/084/SRR15927884/SRR15927884_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/085/SRR15927885/SRR15927885_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/085/SRR15927885/SRR15927885_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/086/SRR15927886/SRR15927886_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/086/SRR15927886/SRR15927886_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/087/SRR15927887/SRR15927887_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/087/SRR15927887/SRR15927887_2.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/088/SRR15927888/SRR15927888_1.fastq.gz
fasp.sra.ebi.ac.uk:/vol1/fastq/SRR159/088/SRR15927888/SRR15927888_2.fastq.gz
一个简单的脚本就可以批量下载上面文本文件里面的全部的 fastq 文件啦:
cat fq.txt |while read iddo ascp -QT -l 300m -P33001 \ -i /home/data/bylin/miniconda3/envs/download/etc/asperaweb_id_dsa.openssh \ era-fasp@$id . done
得到如下所示的文件:
$ ls -lh SRR159278*gz|cut -d" " -f 5-3.5G 2月 6 23:23 SRR15927876_1.fastq.gz3.5G 2月 6 23:14 SRR15927876_2.fastq.gz2.6G 2月 6 23:25 SRR15927877_1.fastq.gz2.7G 2月 6 23:17 SRR15927877_2.fastq.gz2.6G 2月 6 23:14 SRR15927878_1.fastq.gz2.6G 2月 6 23:16 SRR15927878_2.fastq.gz2.8G 2月 6 23:19 SRR15927879_1.fastq.gz2.8G 2月 6 23:15 SRR15927879_2.fastq.gz2.4G 2月 6 23:24 SRR15927880_1.fastq.gz2.4G 2月 6 23:20 SRR15927880_2.fastq.gz2.8G 2月 6 23:17 SRR15927881_1.fastq.gz2.9G 2月 6 23:22 SRR15927881_2.fastq.gz2.3G 2月 6 23:21 SRR15927882_1.fastq.gz2.2G 2月 6 23:22 SRR15927882_2.fastq.gz2.4G 2月 6 23:20 SRR15927883_1.fastq.gz2.3G 2月 6 23:15 SRR15927883_2.fastq.gz2.7G 2月 6 23:19 SRR15927884_1.fastq.gz2.6G 2月 6 23:24 SRR15927884_2.fastq.gz2.6G 2月 6 23:25 SRR15927885_1.fastq.gz2.4G 2月 6 23:23 SRR15927885_2.fastq.gz3.4G 2月 6 23:18 SRR15927886_1.fastq.gz3.3G 2月 6 23:22 SRR15927886_2.fastq.gz2.9G 2月 6 23:21 SRR15927887_1.fastq.gz2.9G 2月 6 23:16 SRR15927887_2.fastq.gz2.2G 2月 6 23:18 SRR15927888_1.fastq.gz2.2G 2月 6 23:26 SRR15927888_2.fastq.gz
可以看到,绝大部分样品的数据量都很小,并不是 10x 那样的动辄几十个 G 的文件。
制作 mapfile
按照官网要求,我们要先做一个 mapfile,告诉 celescope 那些文件属于哪些 sample, 必须要有 3 列:
第一列:Fastq 文件前缀 第二列:Fastq 文件目录路径 第三列:样本名称,是所有输出文件的前缀
下面是官网示例:
$cat ./my.mapfilefastq_prefix1 fastq_dir1 sample1fastq_prefix2 fastq_dir2 sample1fastq_prefix3 fastq_dir1 sample2$ls fastq_dir1fastq_prefix1_1.fq.gz fastq_prefix1_2.fq.gzfastq_prefix3_1.fq.gz fastq_prefix3_2.fq.gz$ls fastq_dir2fastq_prefix2_1.fq.gz fastq_prefix2_2.fq.gz
下面是我自己做的 mapfile 文件 :
SRR15927876 /home/data/bylin/PRJNA764023/01download B19T
SRR15927877 /home/data/bylin/PRJNA764023/01download B11T
SRR15927878 /home/data/bylin/PRJNA764023/01download B11T
SRR15927879 /home/data/bylin/PRJNA764023/01download B9T
SRR15927880 /home/data/bylin/PRJNA764023/01download B9T
SRR15927881 /home/data/bylin/PRJNA764023/01download B9T
SRR15927882 /home/data/bylin/PRJNA764023/01download B4T
SRR15927883 /home/data/bylin/PRJNA764023/01download B4T
SRR15927884 /home/data/bylin/PRJNA764023/01download B13T
SRR15927885 /home/data/bylin/PRJNA764023/01download B13T
SRR15927886 /home/data/bylin/PRJNA764023/01download B19T
SRR15927887 /home/data/bylin/PRJNA764023/01download B2T
SRR15927888 /home/data/bylin/PRJNA764023/01download B2T
可以看到, 其实是 6 个样品,但是每个样品会有多个测序数据。
然后运行下面的代码,会产生每个样本的. sh 文件
conda activate celescopemulti_rna \ --mapfile ./rna.mapfile\###mapfile文件 --genomeDir /home/data/bylin/hs_ensembl_99\####参考基因组位置 --thread 8\###运行线程数 --mod shell###输出的文件格式
这个 multi_rna 命令并不会耗费计算资源,因为它就是帮你输出每个样品一个 shell 脚本,后面仍然是需要运行 shell 脚本的。
打开其中一个 shell 脚本 文件看一下里面有什么
celescope rna sample --outdir .//B11T/00.sample --sample B11T
celescope rna barcode --outdir .//B11T/01.barcode --sample B11T
celescope rna cutadapt --outdir .//B11T/02.cutadapt --sample B11T
celescope rna star --outdir .//B11T/03.star --sample B11T
celescope rna featureCounts --outdir .//B11T/04.featureCounts --sample B11T
celescope rna count --outdir .//B11T/05.count --sample B11T
celescope rna analysis --outdir .//B11T/06.analysis --sample B11T
蛮简单的, 是一个普通的 star 加上 featureCounts 的定量流程。可以看到,就是 celescope 软件的 multi_rna 目录自动根据每个样品的 fastq 文件去产生的运行比对流程的命令,然后运行这些脚本。
批量提交每个样品的 shell 脚本
因为这个时候,其实就是 6 个病人的数据,所以每个病人的 8 个线程, 还是在我的服务器承受范围内。
nohup sh ./shell/*.sh &
官网还特别强调了:必须在工作目录下运行,不应该在 shell 目录下运行它们。(应该是考虑到很多 shell 脚本不熟悉的小伙伴)
要注意的一个问题,那就是不同时间发表的文章,运用的试剂可能是不一样的,官方教程里面的脚本默认是 2 代以上的(scopeV2),但是这篇文章用的是 1 代的试剂,所以在前面设置的时候,要指定试剂,否则会报错:
multi_rna\ --mapfile ./rna.mapfile\ --genomeDir /SGRNJ/Public/Database/genome/homo_mus\ --thread 8\ --mod shell --chemistry scopeV1###在这里指定试剂
其他的具体参数详见官方教程:https://github.com/singleron-RD/CeleScope/blob/master/docs/rna/multi_rna.md
比对结果
比对成功后,会产生对应 sample 的结果,放在对应的文件夹里面
$ ls B11T/
00.sample 02.cutadapt 04.featureCounts 06.analysis
01.barcode 03.star 05.count B11T_report.html
同样的,他会对每个样品产生相应的矩阵和流程报告,跟 10x 类似,供下游 seurat 分析的数据在每个样品的 05.count/B11T_matrix_10X 文件夹里面 ;
$ ls B11T/05.count/*
B11T/05.count/B11T_count_detail.txt B11T/05.count/B11T_downsample.txt
B11T/05.count/B11T_counts.txt B11T/05.count/stat.txt
B11T/05.count/B11T_all_matrix:
barcodes.tsv genes.tsv matrix.mtx
B11T/05.count/B11T_matrix_10X:
barcodes.tsv genes.tsv matrix.mtx
可以看到,这个时候的 genes.tsv 还是比较古老的文件名字。
总体来说,报表也是很容易理解:

报表也是很容易理解
对这个数据集的 6 个病人,进行降维聚类分群和生物学命名也很合理:
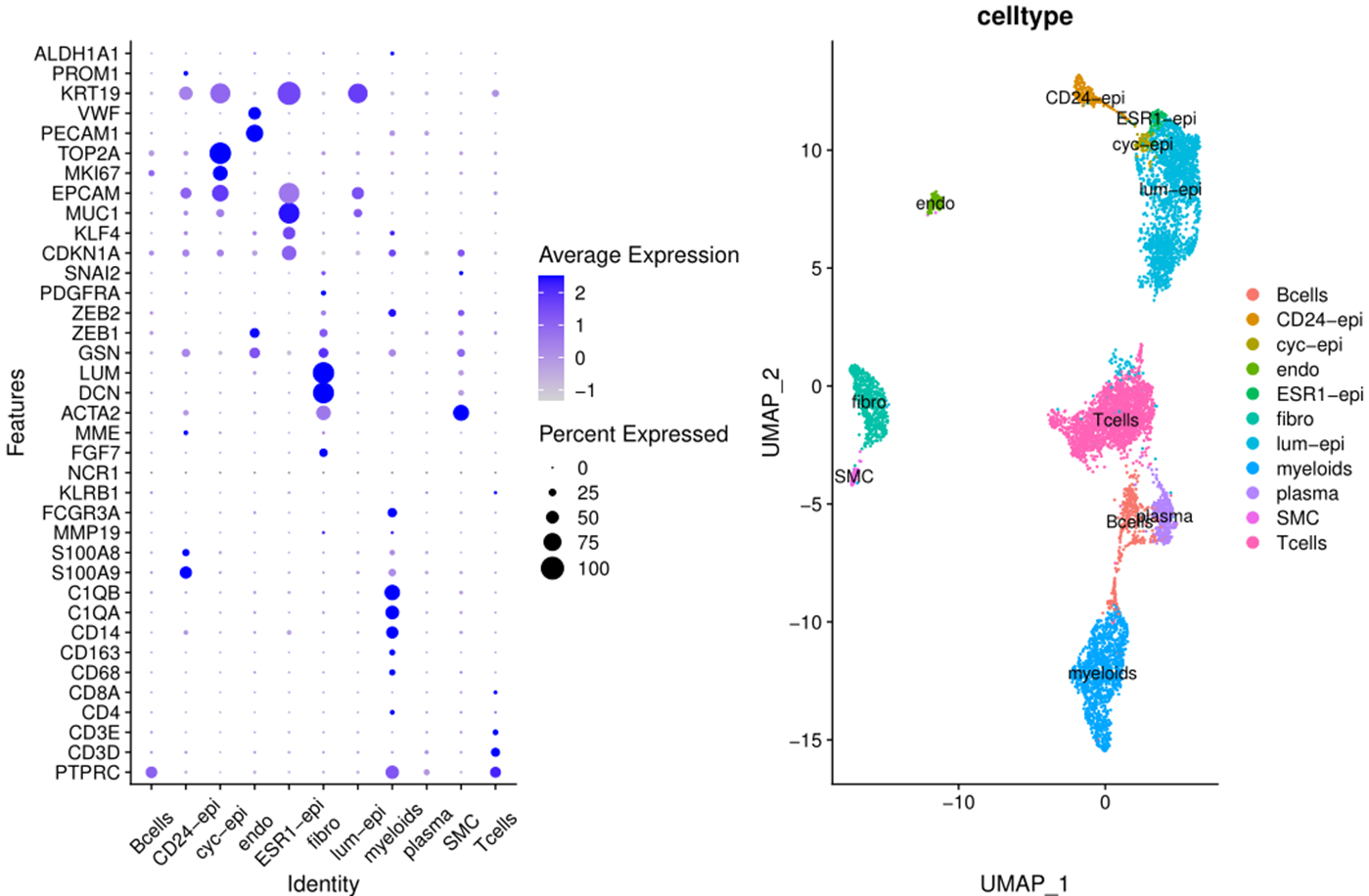
降维聚类分群和生物学命名
转载自《生信技能树》,如有侵权,请联系删除。

