MEGENA(多尺度嵌入基因共表达网络分析)
============================================
 image-20220223204244635
image-20220223204244635
这个网络是由来自伊坎西奈山医学院的科学家们开发的,并于2015年发表《PLoS Computational Biology》中。目前已应用于多种疾病如癌症、肥胖,阿尔茨海默氏症。测试数据来自癌症基因组图谱(TCGA),并且MEGENA在乳腺癌和肺癌中鉴定了新的监管目标,优于其它共表达分析方法。
其实虽然两者也是根据基因-基因相关性分成模块,求与疾病最相关的模块出来,进而筛选关键基因。不过两者肯定是用不同的统计学方法,有兴趣的小伙伴可以比较以下。
Multiscale Embedded Gene Co-expression Network Analysis (plos.org)
MEGENA.pdf (r-project.org)
MEGENA_pipeline (r-project.org)
下面介绍下MEGENA包的主要用法
1. 前言
MEGENA算法的一个关键部分是构造平面滤波网络(PFN)。包括三个主要步骤:1)根据基因表达谱进行相关性分析,筛选出关键基因对构建平面滤波网络(PFN);2)从PFN开始,对模块间进行多尺度聚类分析,得到层次聚类结果;3)采用多尺度中心分析(MHA)来识别高度复杂的网络聚类特征(CTA),探讨模块与临床结果的相关性。
图1显示了MEGENA的总体分析流程。
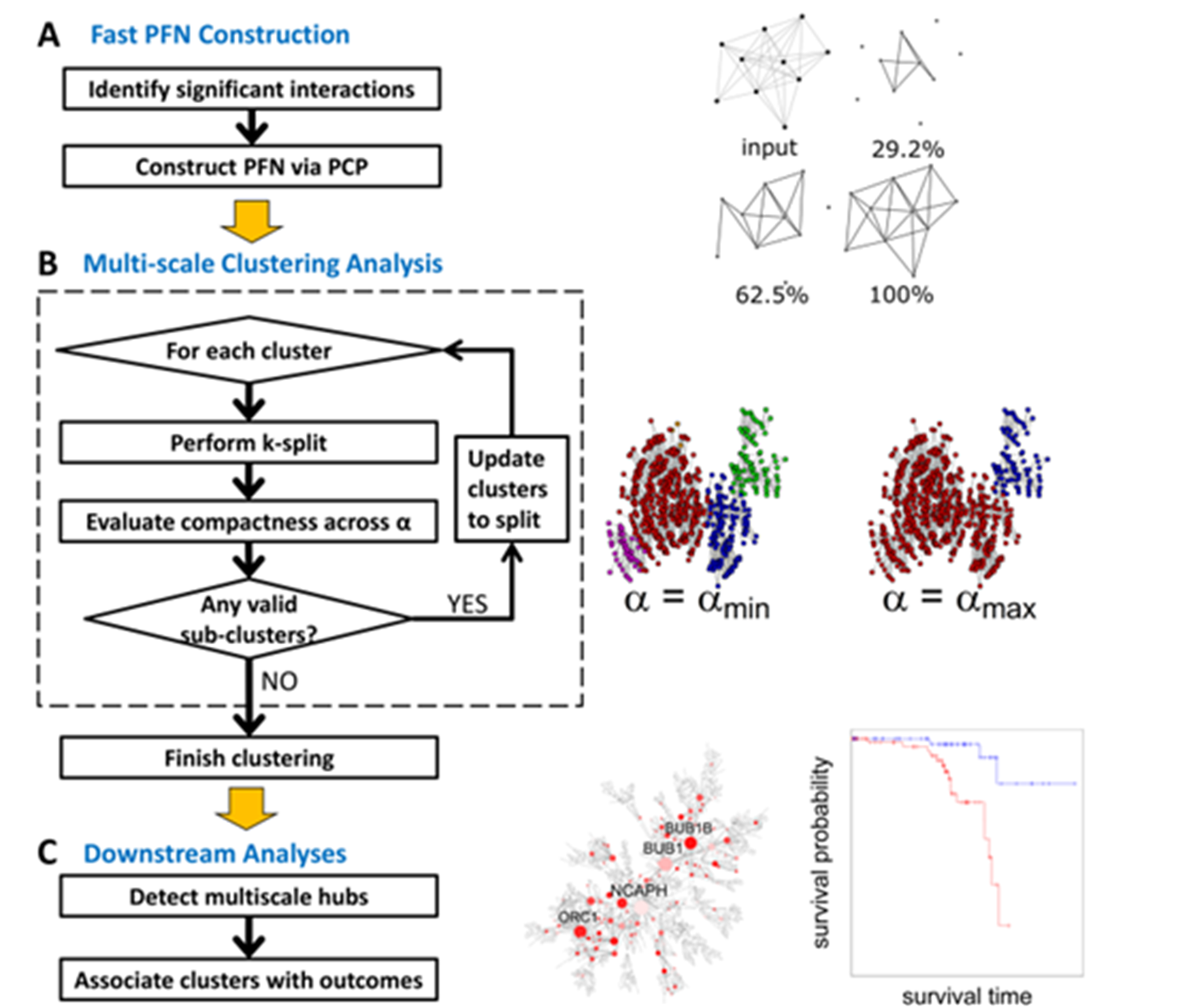 Untitled
Untitled
1. R包安装
rm(list = ls()) # rm R working space# library(devtools);# install_github("songw01/MEGENA");# BiocManager::install('MEGENA')library(MEGENA)
1. 数据载入
• 数据集:datExpr
• A portion of TCGA breast cancer data to test run MEGENA(a gene expression matrix.)
# load toy example datadata(Sample_Expression) dim(datExpr) # TCGA breast cancer data,a gene expression matrixdatExpr[1:4,1:4]
> dim(datExpr)[1] 300 844> datExpr[1:4,1:4] TCGA-E9-A1RC-01A-11R-A157-07 TCGA-BH-A0B1-01A-12R-A056-07C1QB 7.799198 9.491122CD3E 2.703622 6.570632CD3D 1.121214 4.802647TMEM176A 6.608023 8.948846 TCGA-B6-A0RG-01A-11R-A056-07 TCGA-D8-A1JH-01A-11R-A13Q-07C1QB 9.959445 10.648426CD3E 5.812081 8.074163CD3D 4.104806 6.152895TMEM176A 10.054326 8.760917
1. 计算基因表达谱之间的相似性
• 函数:calculate.correlation
• correlation analysis with FDR calculation
原理:用错误发现率(fdr)筛选基因相似性,以尽量减少错误阳性的影响。使用平面滤波网络(PFN)计算有显著相关性的基因对,在使用不同相似度度量的情况下,可以独立地将结果格式化为3列的数据帧,列名为c(row, col, weight),并确保权重列在0到1之间
# input parameters参数n.cores <- 2; #内核/线程数doPar <-TRUE; #是否需要并行method = "pearson" #计算相关性的方法pearson or spearman. FDR.cutoff = 0.05 # FDR阈值,定义相关性module.pval = 0.05 #模块显著性,默认0.05. hub.pval = 0.05 #随机四面体网络连接度p值cor.perm = 10; # number of permutations for calculating FDRs for all correlation pairs. hub.perm = 100; # number of permutations for calculating connectivity significance p-value.# correlation analysis with FDR calculationijw <- calculate.correlation(datExpr, doPerm = cor.perm, output.corTable = FALSE, #输出表格 output.permFDR = FALSE)head(ijw)
## i = 1## i = 2## i = 3## i = 4## i = 5## i = 6## i = 7## i = 8## i = 9## i = 10
> head(ijw) row col rhoC1QB.127 C1QB C1QC 0.9793833CD3E.6 CD3E CD2 0.9793832CD3D.5 CD3D CD2 0.9745410C1QB.3 C1QB C1QA 0.9734056CD3E CD3E CD3D 0.9730448C1QA.123 C1QA C1QC 0.9628771
1. 构建PFN
• 函数:calculate.PFN
• main function to calculate PFN a ranked list of edge pairs
##### calculate PFNel <- calculate.PFN(ijw[,1:3], doPar = doPar,#并行 num.cores = n.cores, #核数 keep.track = FALSE) #创建pfg_el.RData
## ####### PFN Calculation commences ########## [1] "343 out of 894 has been included."## [1] "471 out of 894 has been included."## [1] "556 out of 894 has been included." ## [1] "PFG is complete."
g <- graph.data.frame(el,directed = FALSE) #转化为图数据结构# directed = FALSE为无向网络
5.MCA聚类
• 函数:do.MEGENA
• multiscale clustering analysis (MCA) and multiscale hub analysis (MHA) pipeline
通过MCA聚类进行多尺度聚类分析。“MEGENA.output “是用于下游分析的核心输出,用于汇总和绘图
##### perform MCA clustering.MEGENA.output <- do.MEGENA(g, mod.pval = module.pval, #模块显著性,默认0.05. hub.pval = hub.pval,#随机四面体网络连接度p值 remove.unsig = TRUE, #是否移除不显著的cluster min.size = 10,#最小cluster数 max.size = vcount(g)/2, #最大cluster数 doPar = doPar, #并行 num.cores = n.cores, #核数 n.perm = hub.perm, #计算连接度P值的排列数 save.output = FALSE) #保存每一步分析
1. 汇总结果
• 函数:MEGENA.ModuleSummary
• Summarizes modules into a table
# annotation to be done on the downstreamannot.table=NULLid.col = 1symbol.col= 2###########summary.output <- MEGENA.ModuleSummary(MEGENA.output, mod.pvalue = module.pval,hub.pvalue = hub.pval, min.size = 10,max.size = vcount(g)/2, annot.table = annot.table,id.col = id.col,symbol.col = symbol.col, output.sig = TRUE)
这里要注意annot.table,不过看说明

应该就是节点名≠基因名时,可以再载入个注释表格进行转换,不过指定是哪两列
summary.output$modules
结果得到13个模块
> str(summary.output$modules)List of 13 $ c1_2 : chr [1:133] "C1QB" "C1QA" "SASH3" "HLA-DPA1" ... $ c1_3 : chr [1:121] "CD3E" "CD3D" "CD2" "CXCL11" ... $ c1_4 : chr [1:22] "FCRL5" "CD79A" "LOC96610" "POU2AF1" ... $ c1_5 : chr [1:24] "IFIT3" "IFI44L" "IFIT1" "IFI6" ... $ c1_6 : chr [1:26] "C1QB" "C1QA" "FCGR1A" "FCGR1C" ... $ c1_7 : chr [1:68] "SASH3" "PLEK" "NCKAP1L" "EVI2B" ... $ c1_10: chr [1:65] "CD3E" "CD3D" "CD2" "ITK" ... $ c1_12: chr [1:25] "SLAMF6" "LY9" "GPR174" "CD38" ... $ c1_13: chr [1:11] "TAP1" "HLA-B" "HLA-F" "HLA-H" ... $ c1_15: chr [1:16] "CD79A" "MS4A1" "CD19" "BLK" ... $ c1_19: chr [1:22] "SASH3" "ARHGAP9" "PTPN7" "CD48" ... $ c1_21: chr [1:56] "CD3E" "CD3D" "CD2" "ITK" ... $ c1_23: chr [1:11] "SLAMF6" "LY9" "ZNF831" "BTLA" ...
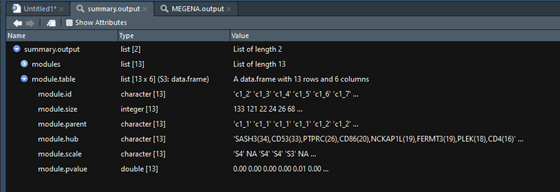
summary.output$module.table
 Untitled
Untitled
若提供了"annot.table",还会出现gene symbol和id对应的结果(mapped.modules)
> print(summary.output$module.table) module.id module.size module.parentc1_2 c1_2 133 c1_1c1_3 c1_3 121 c1_1c1_4 c1_4 22 c1_1c1_5 c1_5 24 c1_1c1_6 c1_6 26 c1_2c1_7 c1_7 68 c1_2c1_10 c1_10 65 c1_3c1_12 c1_12 25 c1_3c1_13 c1_13 11 c1_3c1_15 c1_15 16 c1_4c1_19 c1_19 22 c1_7c1_21 c1_21 56 c1_10c1_23 c1_23 11 c1_12 module.hubc1_2 SASH3(34),CD53(33),PTPRC(26),CD86(20),NCKAP1L(19),FERMT3(19),PLEK(18),CD4(16)c1_3 CD2(33),CD3E(32),GBP5(20),ITK(19),SLAMF6(18)c1_4 MS4A1(12)c1_5 IFIT3(19)c1_6 CD86(14)c1_7 CD53(32),SASH3(27),PLEK(17),NCKAP1L(17),CD4(16)c1_10 CD2(32),CD3E(29)c1_12 SLAMF6(14)c1_13 PSMB9(9)c1_15 MS4A1(12),FCRL1(11)c1_19 SASH3(19),PTPN7(12)c1_21 CD2(32),CD3E(29)c1_23 SLAMF6(10) module.scale module.pvaluec1_2 S4 0.00c1_3 <NA> 0.00c1_4 S4 0.00c1_5 S4 0.00c1_6 S3 0.01c1_7 <NA> 0.00c1_10 <NA> 0.00c1_12 S4 0.00c1_13 S4 0.00c1_15 S3 0.03c1_19 S3 0.00c1_21 S4 0.00c1_23 S3 0.00
1. 可视化
包里还自带绘图函数
pnet.obj <- plot_module(output.summary = summary.output, PFN = g, subset.module = "c1_4", #可视化的cluster layout = "kamada.kawai", #布局的算法:"kamada.kawai"or"fruchterman.reing" label.hubs.only = FALSE, #只展示关键基因的标签 color.code = "grey", output.plot = FALSE, #输出图片 out.dir = "modulePlot", #新建文件夹存结果 col.names = c("magenta","green","cyan"), #网络的颜色 label.scaleFactor = 2, #节点标签大小 hubLabel.col = "black", #标签颜色 hubLabel.sizeProp = 1, #调整关键节点标签与普通的比例,默认0.5 show.topn.hubs = Inf, #显示的最大关键节点数量 show.legend = TRUE) #注释print(pnet.obj[[1]])
 Untitled
Untitled
因为这个网络是有层次结果的,所以输出来的图会呈现出父模块与子模块间的网络关系
不过做出来的图挺丑的,那还不如用老办法,把数据输入到cytoscape做个漂亮的出来
sub_cluster <- summary.output$modules$c1_4 #模块基因#与前面算出权重的el数据集取交集df1 = el[el[,1] %in% sub_cluster,]df2 = df1[df1[,2] %in% sub_cluster,]# df2就可以直接输入cytoscape了,有节点,有边的关系
> head(df2) row col weight22 FCRL5 POU2AF1 0.951707262 CD79A POU2AF1 0.937025497 FCRL5 LOC96610 0.9295689105 FCRL5 CD79A 0.9275319129 LOC96610 TNFRSF17 0.9230158138 FCRL5 TNFRSF17 0.9218706
1. 关键还是得多看文献
不过具体MEGENA网络的用法,如何结合WGCNA网络挖掘关键生物信息呢?关键还是得多看文献哦!
下面近五年用了MEGENA网络方法的高分文章,可以从文献中找到答案哦
Quiescent stem cell marker genes in glioma gene networks are sufficient to distinguish between normal and glioblastoma (GBM) samples | Scientific Reports (nature.com)
Network models of primary melanoma microenvironments identify key melanoma regulators underlying prognosis | Nature Communications
Multiscale network analysis reveals molecular mechanisms and key regulators of the tumor microenvironment in gastric cancer - Song - 2020 - International Journal of Cancer - Wiley Online Library
Network models of prostate cancer immune microenvironments identify ROMO1 as heterogeneity and prognostic marker | Scientific Reports (nature.com)
转载自《生信技能树》,如有侵权,请联系删除。

