使用R包RTCGA把TCGA数据本地化。最新的数据版本是2016-01-28的,本来是可以选择以下的包(但是这些包都被移除了,并不在bioconductor官网啦):
• RTCGA.mutations.20160128
• RTCGA.rnaseq.20160128
• RTCGA.clinical.20160128
• RTCGA.mRNA.20160128
• RTCGA.miRNASeq.20160128
• RTCGA.RPPA.20160128
• RTCGA.CNV.20160128
• RTCGA.methylation.20160128
本来是说旧版本已经可以考虑弃用了,但是因为上面的新版数据包都不存在了,我们反而是只能说退而求其次选择旧版数据。不过TCGA数据其实是2005就开始了,哪怕是截止到2015-11-01版本也不会过时很多。
下面是基于2015-11-01版本的TCGA数据,如果你使用默认安装方法,通常是安装的旧版数据:
• RTCGA.mutations
• RTCGA.rnaseq #测序表达量矩阵
• RTCGA.clinical
• RTCGA.PANCAN12
• RTCGA.mRNA #芯片表达量矩阵
• RTCGA.miRNASeq
• RTCGA.RPPA
• RTCGA.CNV
• RTCGA.methylation
每一个包都是正规的bioconductor包,使用标准安装代码即可:
if (!require("BiocManager", quietly = TRUE)) install.packages("BiocManager")BiocManager::install("RTCGA")BiocManager::install("RTCGA.clinical")BiocManager::install("RTCGA.mRNA")
这个包不仅仅是提供数据整理和下载,也有几个好用的打包的函数,包括:
expressionsTCGA Gather Expressions for TCGA DatasetsmutationsTCGA Gather Mutations for TCGA DatasetsheatmapTCGA Create Heatmaps for TCGA Datasets kmTCGA Plot Kaplan-Meier Estimates of Survival Curves for Survival DatapcaTCGA Plot Two Main Components of Principal Component Analysis
其中expressionsTCGA和mutationsTCGA是从本地R包里面获取表达量信息,另外的3个函数(heatmapTCGA,kmTCGA,pcaTCGA)出图都还凑合,后面我们会一一介绍。
支持的数据集
最主要的包RTCGA有一个函数infoTCGA可以查询他们团队整理的数据集,其实就是https://gdac.broadinstitute.org/网页里面的数据集:
library(RTCGA) all_TCGA_cancers=infoTCGA()DT::datatable(all_TCGA_cancers)
如下所示:

支持的数据集
既然也是https://gdac.broadinstitute.org/,其实就跟通过R包RTCGAToolbox链接FireBrowse来探索TCGA等公共数据类似了,因为都是broad研究所的整理。
首先需要使用expressionsTCGA和mutationsTCGA从本地R包里面获取表达量信息,然后使用前面提到了有3个函数(heatmapTCGA,kmTCGA,pcaTCGA)可以进行统计可视化,我们就来逐一介绍。
expressionsTCGA
首先我们需要对任意基因从任意癌症里面获取芯片表达数据,这里我们拿下面3种癌症做示范:
• Breast invasive carcinoma (BRCA)
• Ovarian serous cystadenocarcinoma (OV)
• Lung squamous cell carcinoma (LUSC)
我们拿一些免疫基因作为例子,其中要注意的是mRNA并不是rnaseq两者不太一样,具体样本数量,可以看最前面的表格。
library(RTCGA)library(RTCGA.mRNA)cg=c('CD3D','CD3G','CD247','IFNG','IL2RG','IRF1','IRF4','LCK','OAS2','STAT1')cgexpr <- expressionsTCGA(BRCA.mRNA, OV.mRNA, LUSC.mRNA, extract.cols = cg ) exprnb_samples <- table(expr$dataset)nb_sampleslibrary(ggpubr)ggboxplot(expr, x = "dataset", y = "CD3D", title = "CD3D", ylab = "Expression", color = "dataset", palette = "jco", rotate = TRUE)
可以看到在这3个癌症里面CD3D基因的表达量范围是不一样的:
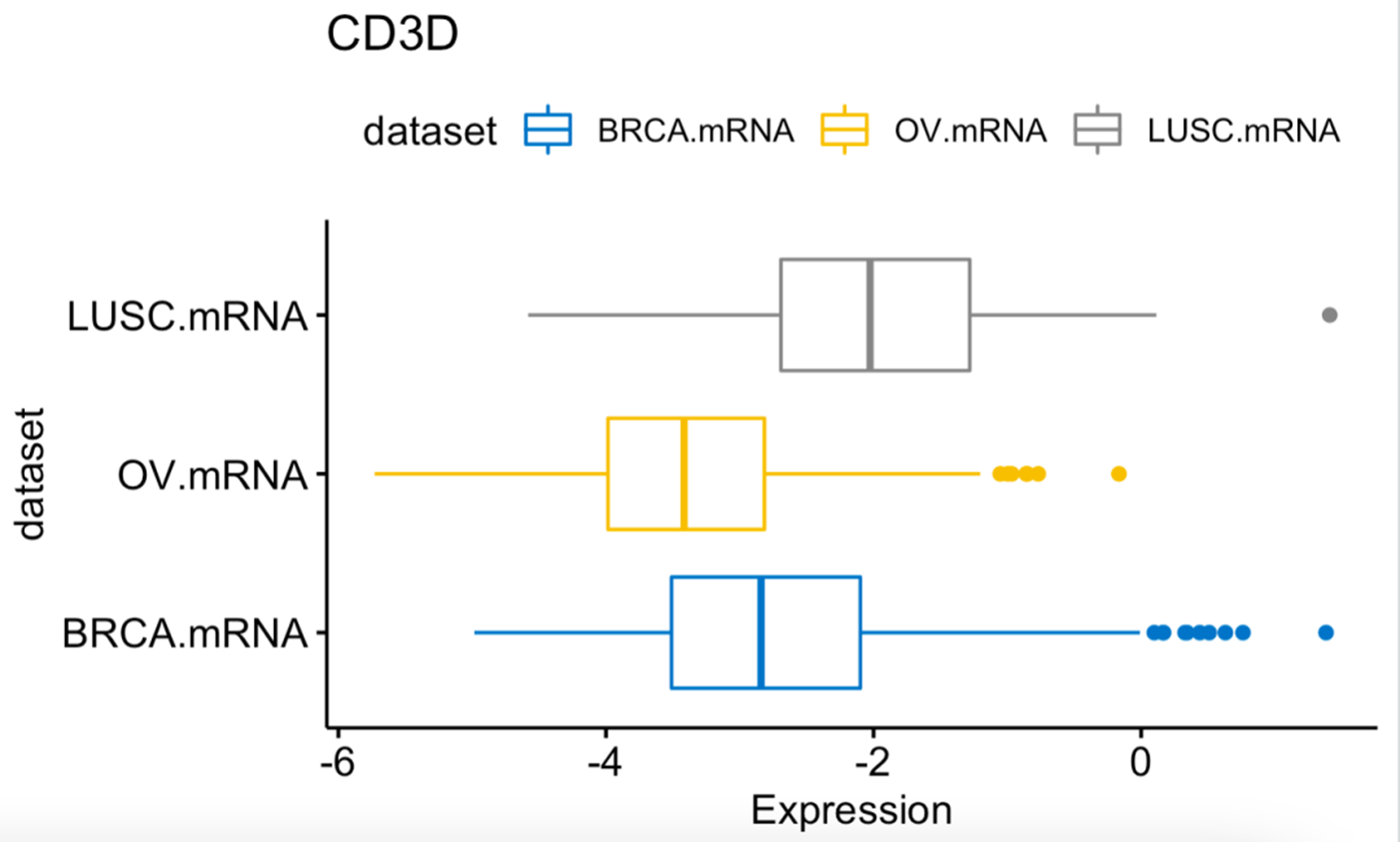
3个癌症里面CD3D基因的表达量范围是不一样
不过,这个数据是mRNA并不是rnaseq,而且看起来是被zscore了的,这样的值在不同数据集里面的对比起来是有问题的。
大家可以安装另外的包,去看看转录组测序数据里面的这些基因的表达量。而且测序数据里面的基因还比较麻烦,不仅仅是要symbol还要entrez的ID,具体需要看https://wiki.nci.nih.gov/display/TCGA/RNASeq+Version+2的解释。
mutationsTCGA
也是同样的安装:
> BiocManager::install('RTCGA.mutations')# RTCGA.mutations_20151101.22.0.tar.gz'# Content type 'application/x-gzip' length 108858817 bytes (103.8 MB)
可以看到仍然是2015-11-01的数据,文件比较大,所以仍然是考验网络速度。
library(RTCGA.mutations) library(dplyr)mutationsTCGA(BRCA.mutations, OV.mutations) %>% filter(Hugo_Symbol == 'TP53') %>% filter(substr(bcr_patient_barcode, 14, 15) == "01") %>% # cancer tissue mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) -> BRCA_OV.mutationstable(BRCA_OV.mutations$dataset)
可以看到,我们很轻松的就从OV和BRCA两个癌症里面,拿到了有TP53突变的病人的ID,可以看到:
> table(BRCA_OV.mutations$dataset)BRCA.mutations OV.mutations 306 279 > as.data.frame(tail(sort(table(BRCA_OV.mutations$Variant_Classification)))) Var1 Freq1 Splice_Site 202 Frame_Shift_Ins 233 Splice_Site_SNP 324 Frame_Shift_Del 645 Nonsense_Mutation 706 Missense_Mutation 352
两个癌症数据集里面的都有几百个病人有TP53基因的突变,而且绝大部分突变类型都是Nonsense_Mutation和Missense_Mutation。
这样的突变信息,就可以对病人进行分组后,看生存差异,我在生信技能树多次分享过生存分析的细节;
• 人人都可以学会生存分析(学徒数据挖掘)
• 学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?
• 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
• 学徒作业-两个基因突变联合看生存效应
heatmapTCGA
我感觉完全没有必要使用它的函数,因为热图基本上大家都是可以自定义,再不济至少可以pheatmap一下。比如前面的3个癌症数据集的表达量矩阵。简单的代码如下所示:
ar=data.frame(group=expr$dataset)rownames(ar)=expr$bcr_patient_barcodehead(ar)ct=expr[,3:ncol(expr)]rownames(ct)=expr$bcr_patient_barcodepheatmap::pheatmap(ct, show_rownames = F, annotation_row = ar)
可以看到,虽然前面3个癌症里面CD3D基因的表达量范围有差异,但是这些免疫集联合起来就没有那么强烈的癌症特异性了,说明免疫这个变量在每个癌症内部都是很具有异质性的,所以不同癌症很难根据免疫进行区分。
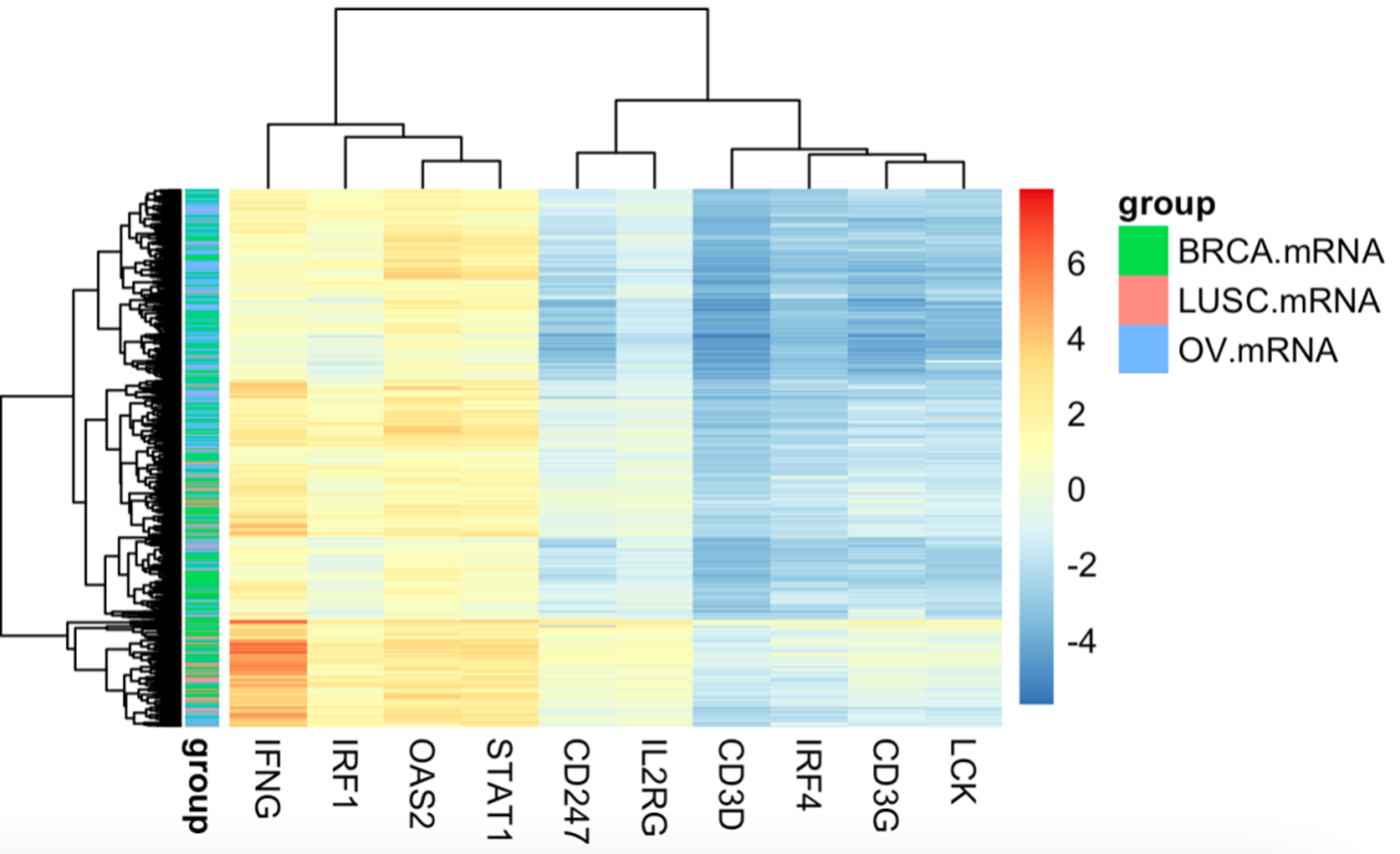
免疫这个变量在每个癌症内部都是很具有异质性
pcaTCGA
前面我们介绍了仅仅是根据指定的基因列表,就可以筛选表达量矩阵,并且进行合理的分组,见:
• 基于基因集的样品队列分组之gsea等打分
• 基于基因集的样品队列分组之层次聚类
• 基于基因集的样品队列分组之PCA
好像也没有必要使用作者的pcaTCGA函数。
kmTCGA
我们前面筛选到了OV和BRCA两个癌症的TP53突变信息,现在就可以结合起来临床信息,使用它的kmTCGA函数,进行生存分析:
library(RTCGA.clinical)survivalTCGA( BRCA.clinical, OV.clinical, extract.cols = "admin.disease_code") %>% dplyr::rename(disease = admin.disease_code) -> BRCA_OV.clinicalBRCA_OV.clinical %>% left_join( BRCA_OV.mutations, by = "bcr_patient_barcode" ) %>% mutate(TP53 = ifelse(!is.na(Variant_Classification), "Mut","WILDorNOINFO")) -> BRCA_OV.clinical_mutationsBRCA_OV.clinical_mutations %>% select(times, patient.vital_status, disease, TP53) -> BRCA_OV.2plotkmTCGA( BRCA_OV.2plot, explanatory.names = c("TP53", "disease"), break.time.by = 400, xlim = c(0,2000), pval = TRUE) -> km_plotkm_plot
如下所示:
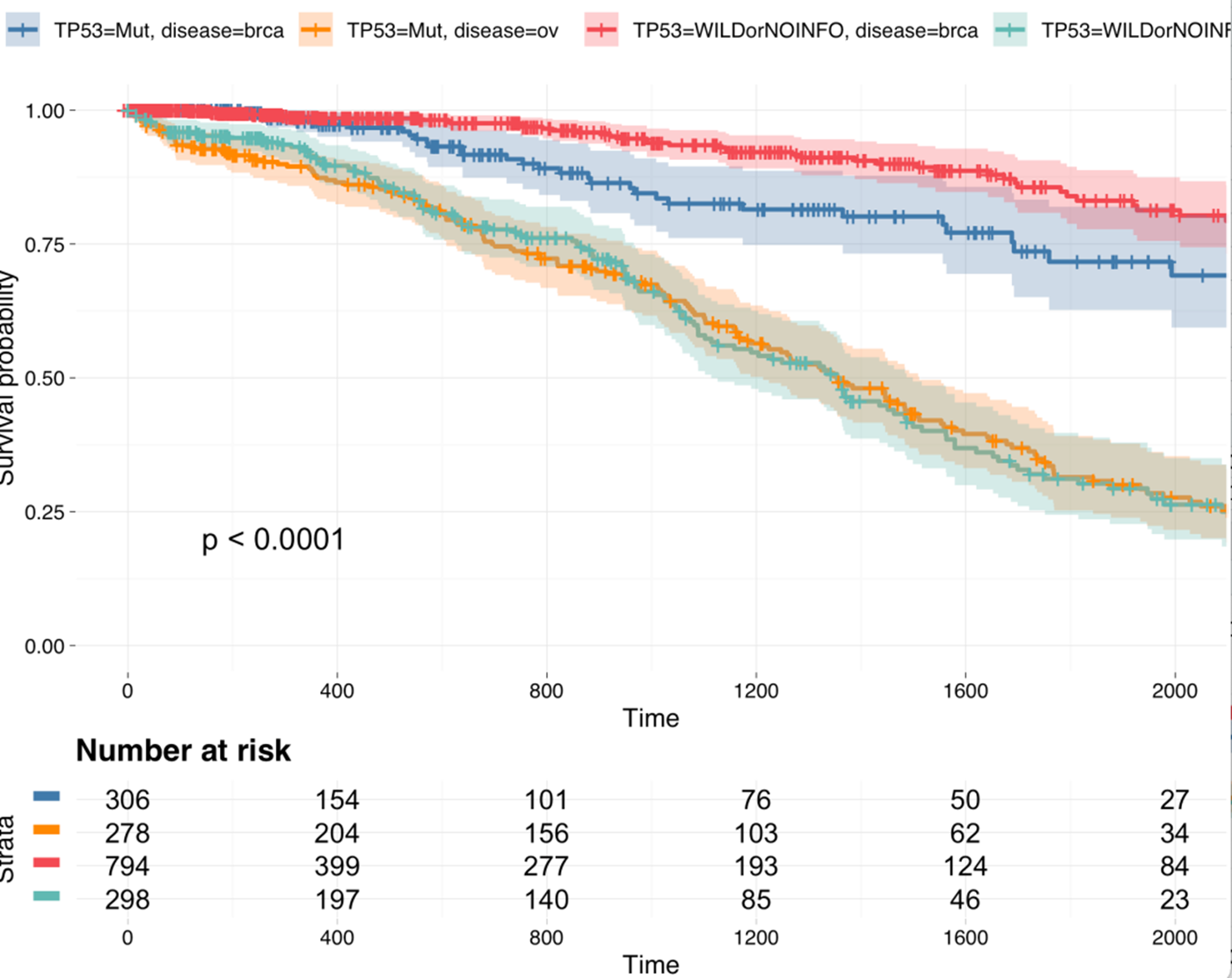
TP53基因突变在两个癌症的生存意义
我在生信技能树多次分享过生存分析的细节;
• 人人都可以学会生存分析(学徒数据挖掘)
• 学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?
• 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
• 学徒作业-两个基因突变联合看生存效应
转载自《生信技能树》,如有侵权,请联系删除。

