TCGA数据最权威的应该是GDC官网入口(https://portal.gdc.cancer.gov/)啦,但是我们前面的几个笔记都没有提到它,见:
• 通过R包cgdsr链接cbioportal来探索TCGA等公共数据
• 通过R包RTCGAToolbox链接FireBrowse来探索TCGA等公共数据
主要是因为GDC官网虽然权威,但是太复杂了,不利于初学者。而且GDC官网是针对TCGA数据库的每个癌症的每个病人的不同数据分开存放,每次都是批量下载后,整理合并的。但是我们前面的在线接口,去cbioportal或者FireBrowse都是以癌症为单位下载不同数据集。包括后面分享的:
• 使用R包RTCGA把TCGA数据本地化
• 使用MultiAssayExperiment结构探索TCGA数据
都是如此,以癌症为单位拿到数据集后,进而去筛选符合要求的病人。相当于是数据框取子集。但是GDC官网里面的数据信息存放单位是样品,每次都是根据要求下载指定的数据即可,无需取子集。
其实GDC官网也是有R包接口,就是TCGA数据库R包集大成者TCGAbiolinks,可以看到其教程非常丰富https://www.bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html
•
1. Introduction
•
1. Searching GDC database
•
1. Downloading and preparing files for analysis
•
1. Clinical data
•
1. Mutation data
•
1. Compilation of TCGA molecular subtypes
•
1. Analyzing and visualizing TCGA data
•
1. Case Studies
•
1. Graphical User Interface (GUI)
•
1. Classifiers
•
1. TCGAbiolinks_Extension
核心功能:指定数据下载
摸索这个TCGAbiolinks包的过程就是对TCGA数据库的从零开始学习过程,它介绍了TCGA数据库的癌症类型,每个癌症有哪些数据形式。这些都是使用TCGAbiolinks包时候所需要指定的参数:
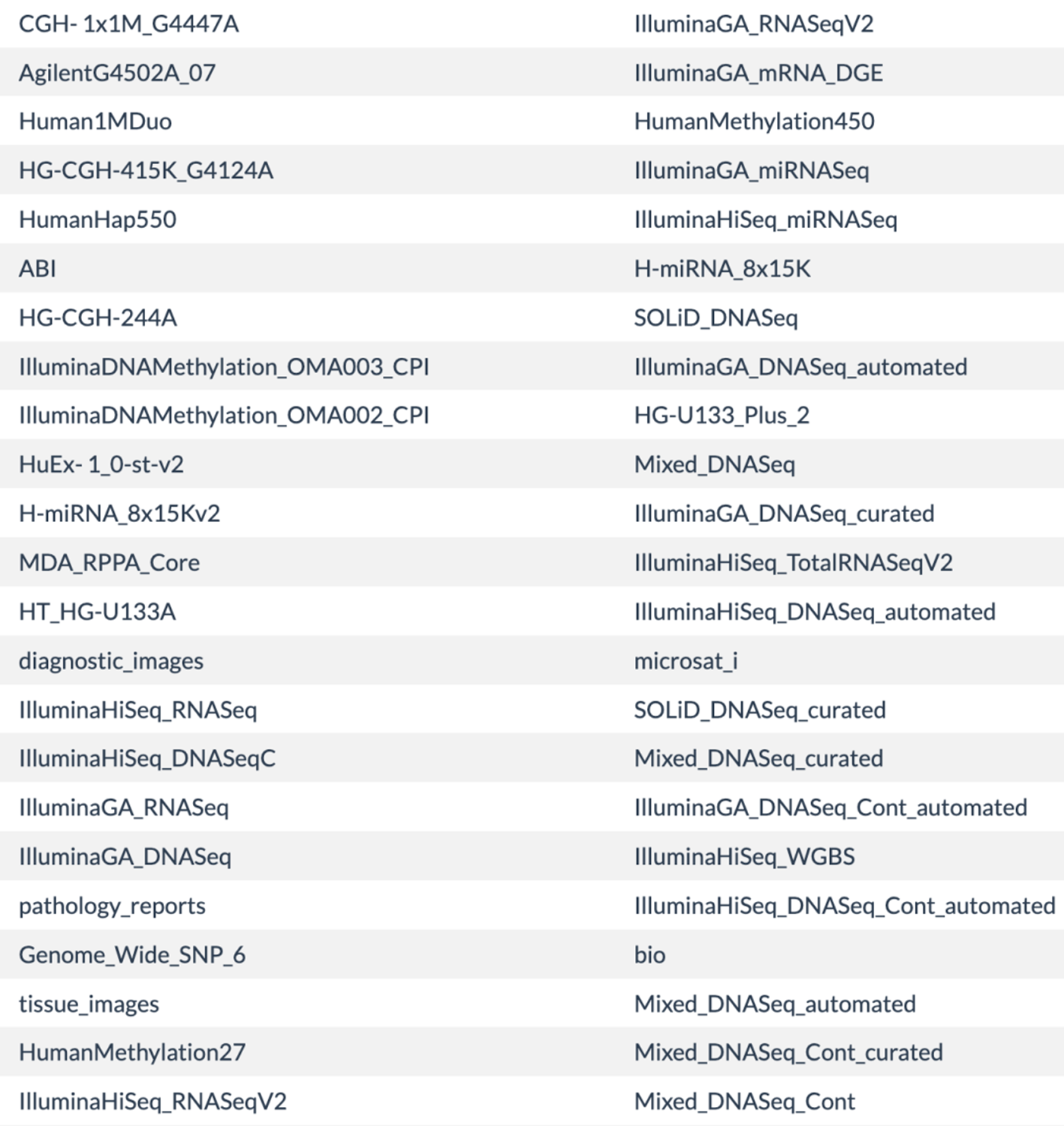
数据形式
参数之详细,基本上很难背诵下来,只能说看着文档,边查询边使用了。https://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/query.html
只需要摸索好了4个参数,就可以组合进行各式各样的数据下载。比如下载两种脑癌的甲基化数据:
query <- GDCquery( project = c("TCGA-GBM", "TCGA-LGG"), data.category = "DNA Methylation", legacy = FALSE, platform = c("Illumina Human Methylation 450"), sample.type = "Recurrent Tumor")
如果是需要下载其它癌症的其它类型数据,变换里面的参数即可。
有了这样的GDCquery函数准备好的query变量,就可以后续使用GDCdownload函数从GDC官网下载,然后使用GDCprepare把下载的文件整理成为一个SummarizedExperiment格式的对象进行后续分析。
因为网络问题,我们没办法那甲基化数据距离,所以这里仍然是选择表达量矩阵,而且仅仅是挑选2个病人:
query.exp.hg19 <- GDCquery( project = "TCGA-GBM", data.category = "Gene expression", data.type = "Gene expression quantification", platform = "Illumina HiSeq", file.type = "normalized_results", experimental.strategy = "RNA-Seq", barcode = c("TCGA-14-0736-02A-01R-2005-01", "TCGA-06-0211-02A-02R-2005-01"), legacy = TRUE)GDCdownload(query.exp.hg19,method = "api")data <- GDCprepare(query.exp.hg19)
还不到1Mb的文件,我下载了六次才成功,真的是伟大的中国长城啊:
> GDCdownload(query.exp.hg19,method = "api")Downloading data for project TCGA-GBMGDCdownload will download 2 files. A total of 873.555 KBDownloading as: Mon_Mar_28_12_51_16_2022.tar.gzDownloading: 370 kB Starting to add information to samples => Add clinical information to samples => Adding TCGA molecular information from marker papers => Information will have prefix 'paper_' gbm subtype information from:doi:10.1016/j.cell.2015.12.028
得到了2个病人的接近2万个基因的表达量矩阵的一个对象,是SummarizedExperiment格式:
> data
class: RangedSummarizedExperiment
dim: 19947 2
metadata(1): data_release
assays(1): normalized_count
rownames(19947): A1BG A2M ... TICAM2 SLC25A5-AS1
rowData names(3): gene_id entrezgene ensembl_gene_id
colnames(2): TCGA-14-0736-02A-01R-2005-01 TCGA-06-0211-02A-02R-2005-01
colData names(105): barcode patient ...
paper_Telomere.length.estimate.in.blood.normal..Kb.
paper_Telomere.length.estimate.in.tumor..Kb.
其它格式的数据下载,大同小异,这里就不再赘述。
突变信息
目前支持3种来源的突变信息,主要是基于hg38和hg19,以及最新的MC3计划整理的突变:
1. UseGDCquery_Mafwhich will download MAF aligned against hg38
2. UseGDCquery,GDCdownloadandGDCpreprareto download MAF aligned against hg19
3. UsegetMC3MAF(), to download MC3 MAF fromhttps://gdc.cancer.gov/about-data/publications/mc3-2017
多年前我就系统性的介绍过:TCGA的pan-caner资料大全(以后挖掘TCGA数据库就用它)还专门指出了癌症的somatic突变的maf文件问题:TCGA数据库maf突变资料官方大全。起初TCGA数据库的全部数据都是提供下载的,包括fastq,bam,vcf,但是呢,后来因为保护病人隐私,就只开放maf格式的somatic突变数据下载。癌症的somatic突变概念需要自行搜索学习,如果你还不了解maf格式,请看:https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/,其实很多包都提供类似的接口,比如TCGAmutations包整合了TCGA中全部样本的maf文件
# devtools::install_github(repo = "PoisonAlien/TCGAmutations") library(TCGAmutations) tmp=as.data.frame(tcga_available())
感兴趣的可以去玩一下,我们现在要介绍的TCGA数据库R包集大成者TCGAbiolinks也是一句话代码下载全部的突变数据,本质上就是MC3计划提供的maf文件下载并且读取而已。
library(SummarizedExperiment)library(TCGAbiolinks)maf <- getMC3MAF()
这个getMC3MAF函数会耗费很长时间下载一个比较大(718.4 MB)的文件:
----------------------------------------------------------------------------------------------------o Starting to download Public MAF from GDCo More information at: https://gdc.cancer.gov/about-data/publications/mc3-2017o Please, cite: Cell Systems. Volume 6 Issue 3: p271-281.e7, 28 March 2018 10.1016/j.cels.2018.03.002试开URL’https://api.gdc.cancer.gov/data/1c8cfe5f-e52d-41ba-94da-f15ea1337efc'Content type 'application/octet-stream' length 753339089 bytes (718.4 MB)
如果不是做泛癌研究,其实可以简简单单使用GDCquery_Maf即可,它支持我们指定癌症肿瘤,这样文件小很多,比如:
library(SummarizedExperiment)library(TCGAbiolinks) library(maftools)library(dplyr)maf <- GDCquery_Maf("CHOL", pipelines = "muse") %>% read.maf plotmafSummary(maf = maf, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE)oncoplot(maf = maf, top = 10, removeNonMutated = TRUE)titv = titv(maf = maf, plot = FALSE, useSyn = TRUE) plotTiTv(res = titv)
一下子可以出很多图,感兴趣的可以去自行阅读maftools的文档,比如突变全景图,如下所示:
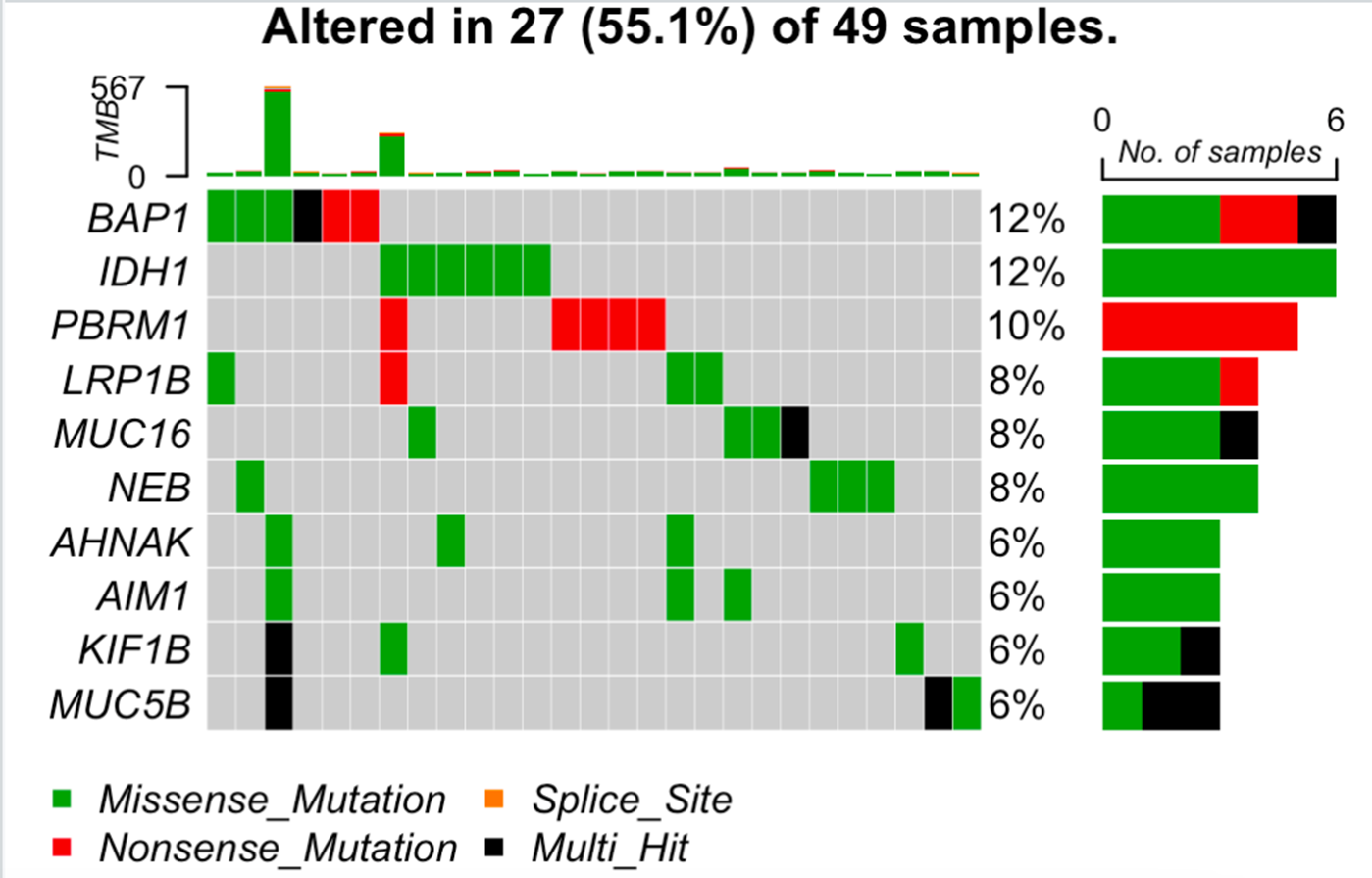
突变全景图
因为对肿瘤样品找somatic突变的数据分析流程很多,所以还有一个pipelines参数可以调整,供选择的是:muse,varscan2,somaticsniper,mutect.
癌症的somatic突变概念需要自行搜索学习,如果你还不了解maf格式,请看:https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/
癌症的分子分型
众所周知,TCGA数据库是目前最综合全面的癌症病人相关组学数据库,包括的多组学数据有:
• DNA Sequencing (WGS/WES)
• mRNA/miRNA Sequencing
• Protein Expression Array
• Array-based Expression
• DNA Methylation Array
• Copy Number Array
而TCGA数据库的全部的癌症列表如下:
ACC - Adrenocortical carcinoma
BLCA - Bladder Urothelial Carcinoma
BRCA - Breast invasive carcinoma
CESC - Cervical and endocervical cancers
CHOL - Cholangiocarcinoma
COAD - Colon adenocarcinoma
COADREAD - Colorectal adenocarcinoma
DLBC - Diffuse Large B-cell Lymphoma
ESCA- Esophageal carcinoma
GBM - Glioblastoma multiforme
GBMLGG - GBMLGG-Glioma
HNSC - Head and Neck squamous cell carcinoma
KICH - Kidney Chromophobe
KIPAN - Pan-kidney cohort (KICH+KIRC+KIRP)
KIRC - Kidney renal clear cell carcinoma
KIRP - Kidney renal papillary cell carcinoma
LAML - Acute Myeloid Leukemia
LGG - Brain Lower Grade Glioma
LIHC - Liver hepatocellular carcinoma
LUAD - Lung adenocarcinoma
LUSC - Lung squamous cell carcinoma
MESO - Mesothelioma
OV - Ovarian serous cystadenocarcinoma
PAAD - Pancreatic adenocarcinoma
PCPG - Pheochromocytoma and Paraganglioma
PRAD - Prostate adenocarcinoma
READ - Rectum adenocarcinoma
SARC - Sarcoma
SKCM - Skin Cutaneous Melanoma
STAD - Stomach adenocarcinoma
STES - Stomach and Esophageal carcinoma
TGCT - Testicular Germ Cell Tumors
THCA - Thyroid carcinoma
THYM - Thymoma
UCEC - Uterine Corpus Endometrial Carcinoma
UCS - Uterine Carcinosarcoma
UVM - Uveal Melanoma
上面的分类是常规组织器官来源分类方式,其实癌症的异质性已经是广为人知了,基本上TCGA计划每次对一个癌症的多组学进行总结和整理的时候,就会依据表达量模式以及其它组学特性把成百上千个癌症病人划分为三五个有固定模式的分子分型。
比如乳腺癌,你可以看lumA,lumB,basal,HER2等亚型,其中TNBC可以继续细分为3~7种亚型,当然了,现在有了单细胞转录组数据的加持,细胞亚型会越来越清晰。如果要整合多组学数据,分类也会更加复杂。
再比如是胃癌,也是有4种分子分型,具体如下:
• ①爱泼斯坦-巴尔(Epstein-Barr)病毒(EBV)阳性型肿瘤:约占胃癌的9%,表现为较高频率的PIK3CA基因突变和DNA极度超甲基化,以及JAK2、CD274(也称PD-L1)和PDCD1LG2(也称PD-L2)基因扩增。
• ②微卫星不稳定(MSI)型:约占22%,表现为重复DNA序列突变增加,包括编码靶向致癌信号蛋白的基因突变。
• ③基因稳定(GS)型:约占20%,其组织学变异弥漫且丰富,RHOA基因突变或RHO家族GTP酶活化蛋白基因融合现象多见。
• ④染色体不稳定(CIN)型:此类肿瘤占胃癌的比例近一半,表现为显著异倍体性及受体酪氨酸激酶的局部扩增。
这些分子分型信息,在TCGAbiolinks也有,使用PanCancerAtlas_subtypes可以获取全部的不同癌症的不同分子分型,使用TCGAquery_subtype可以获取指定癌症的分子分型信息。
library(TCGAbiolinks)library(SummarizedExperiment)library(dplyr)library(DT) subtypes <- PanCancerAtlas_subtypes() lgg.gbm.subtype <- TCGAquery_subtype(tumor = "lgg")
这个PanCancerAtlas_subtypes函数,会对目前有定论的七千多个肿瘤样品(TCGA数据库有超一万的肿瘤样品哦)返回其分子分型信息。如果一个癌症没有达成统一共识,就会从点突变,拷贝数,mRNA表达量,miRNA表达量,甲基化等多个角度进行分子分型,选择相对流行的分子分型给大家。对乳腺癌这样的研究比较多的癌症,就有共识了,只有PMA50分子分型,就是lumA,lumB,basal,HER2等亚型。
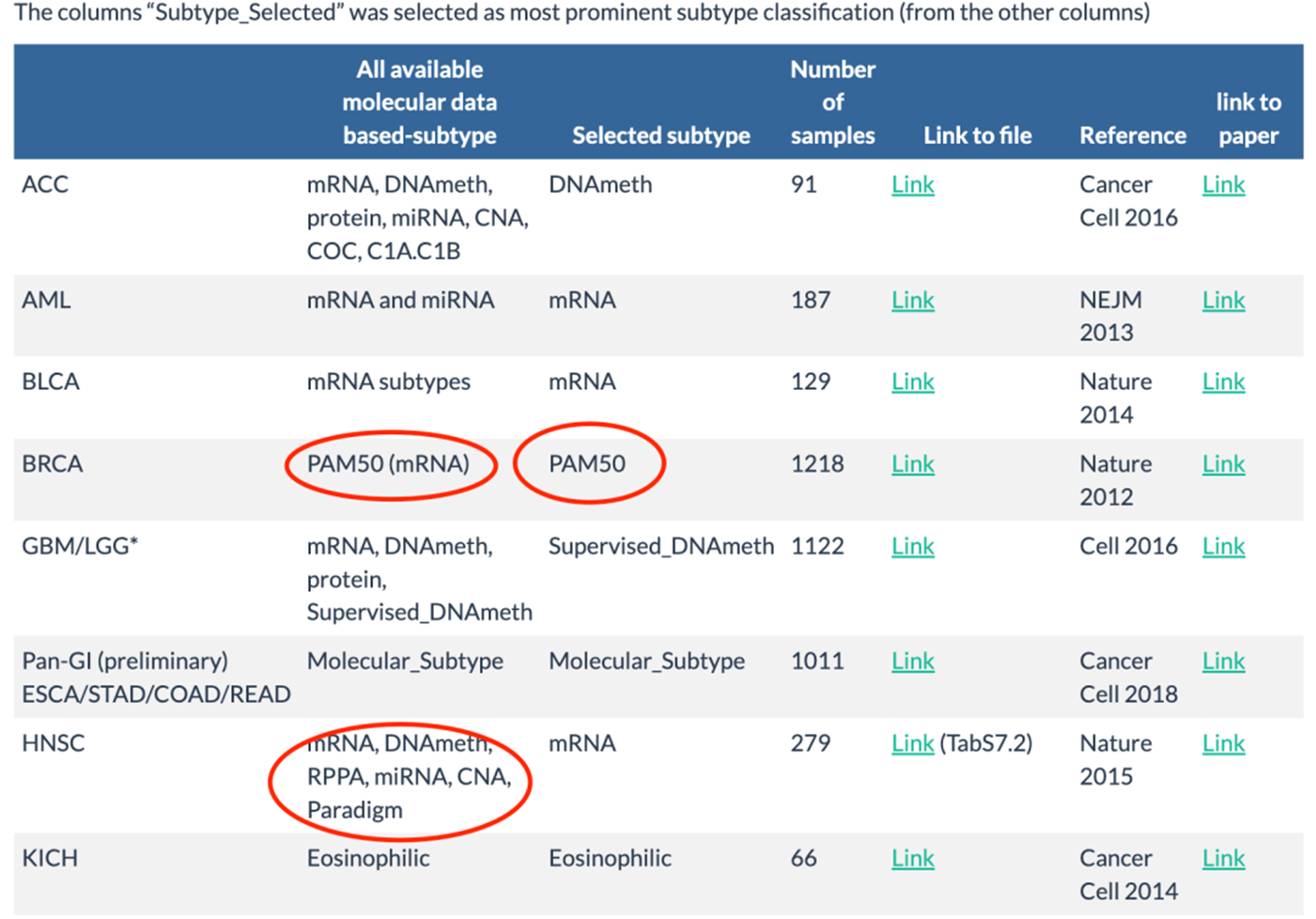
而且TCGAquery_subtype是针对指定癌症,所以它里面的信息不一样,基本上是依据不同的肿瘤数据集对应的文献的分子分型整理。
分析流程
参考文档:https://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/analysis.html
数据分析函数:
数据分析的前提是有数据,而前面我们演示了可以使用TCGAbiolinks进行任意癌症的任意数据下载,都是SummarizedExperiment格式的对象,它无缝连接给TCGAbiolinks包里面的数据分析函数,主要是差异分析和生存分析啦:
它这个包的帮助文档里面列出来的函数超级多,不过官方文档里面仅仅是讲解了如下所示:
• TCGAanalyze_Preprocessing: Preprocessing of Gene Expression data (IlluminaHiSeq_RNASeqV2)
• TCGAanalyze_DEA&TCGAanalyze_LevelTab: Differential expression analysis (DEA)
• TCGAanalyze_EAcomplete & TCGAvisualize_EAbarplot: Enrichment Analysis
• TCGAanalyze_survival: Survival Analysis
• TCGAanalyze_SurvivalKM: Correlating gene expression and Survival Analysis
• TCGAanalyze_DMR: Differentially methylated regions Analysis
当然了,大家可以无需理会这些函数,因为本质上它也是处理一个转录组测序表达量矩阵或者是一个甲基化矩阵,我们针对常规的ngs组学其实是有自己的数据分析流程的,它这些包装好的函数跟我们自己的写是一回事。

一系列数据分析函数
这里面的函数每一个都是独立的知识点,后续我们的这个专辑会一一介绍,大家可以先看看里面的图表!目前简单的差异分析流程,基本上转录组测序技术和芯片技术拿到的表达量矩阵后续分析大同小异,公众号推文在:
• 解读GEO数据存放规律及下载,一文就够
• 解读SRA数据库规律一文就够
• 从GEO数据库下载得到表达矩阵一文就够
• GSEA分析一文就够(单机版+ R语言版)
• 根据分组信息做差异分析-这个一文不够的
• 差异分析得到的结果注释一文就够
我也在生信技能树多次分享过生存分析的细节;
• 人人都可以学会生存分析(学徒数据挖掘)
• 学徒数据挖掘之谁说生存分析一定要按照表达量中位值或者平均值分组呢?
• 基因表达量高低分组的cox和连续变量cox回归计算的HR值差异太大?
• 学徒作业-两个基因突变联合看生存效应
可视化函数
它这个包的帮助文档里面列出来的函数超级多,不过官方文档里面仅仅是讲解了如下所示:
• TCGAvisualize_Heatmap: Create heatmaps with cluster bars
• TCGAvisualize_Volcano: Create volcano plot
• TCGAvisualize_PCA: Principal Component Analysis plot for differentially expressed genes
• TCGAvisualize_meanMethylation: Mean DNA Methylation Analysis
• TCGAvisualize_starburst: Integration of gene expression and DNA methylation data
当然了,大家可以无需理会这些函数,绘图而已,它这些包装好的函数跟我们自己的写是一回事。

一系列绘图函数
只需要学会R基础,绘图都是慢工出细活。
4个案例
参考文档:https://www.bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/casestudy.html
• Case study 1: Pan Cancer downstream analysis BRCA
• Case study 2: Pan Cancer downstream analysis LGG
• Case study 3: Integration of methylation and expression for ACC
• Case study 4: Elmer pipeline - KIRC
这4个案例,相当于是把前面的指定基因或者癌症去下载TCGA数据库文件,以及各式各样的分析和可视化步骤串联起来了。
当然了,因为这个TCGAbiolinks包的出名,也有很多其它团队给出自己的实例,比如:https://cran.r-project.org/web/packages/GeoTcgaData/readme/README.html
扩展:分子分型模型的应用
可能是开发这个TCGA数据库R包集大成者TCGAbiolinks工具的团队背景是脑癌,我看它总是拿LGG和GBM举例,还专门开发一个函数gliomaClassifier,可以根据甲基化芯片数据,比如27K,450K,850K,对一个病人进行分子分型预测,包括下面的6个分型:
• Mesenchymal-like
• Classic-like
• G-CIMP-high
• G-CIMP-low
• LGm6-GBM
• Codel
这个模型参考文献是:“Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma.” Cell 164.3 (2016): 550-563.(https://doi.org/10.1016/j.cell.2015.12.028)
我看了看它这个包的帮助文档,里面并没有其它癌症的其它组学的分子分型模型预测函数,不过道理是相通的。我们后续会讲解不同的预测模型及其R代码实现方式。
转载自《生信技能树》,如有侵权,请联系删除。

