用于实战的数据集来自下面这篇于2017年发表在The Plant Journal的文章《Different mutational function of low- and high-linear energy transfer heavy-ion irradiation demonstrated by whole-genome resequencing of Arabidopsis mutants》
分析用到的软件
• sratoolkits
• fastp
• samtools
• bwa
• GATK、picard
(1)原始测序数据&参考基因组下载&索引构建
首先根据文章的Bioproject编号(PRJDB5412),找到SRA Experiments这一栏
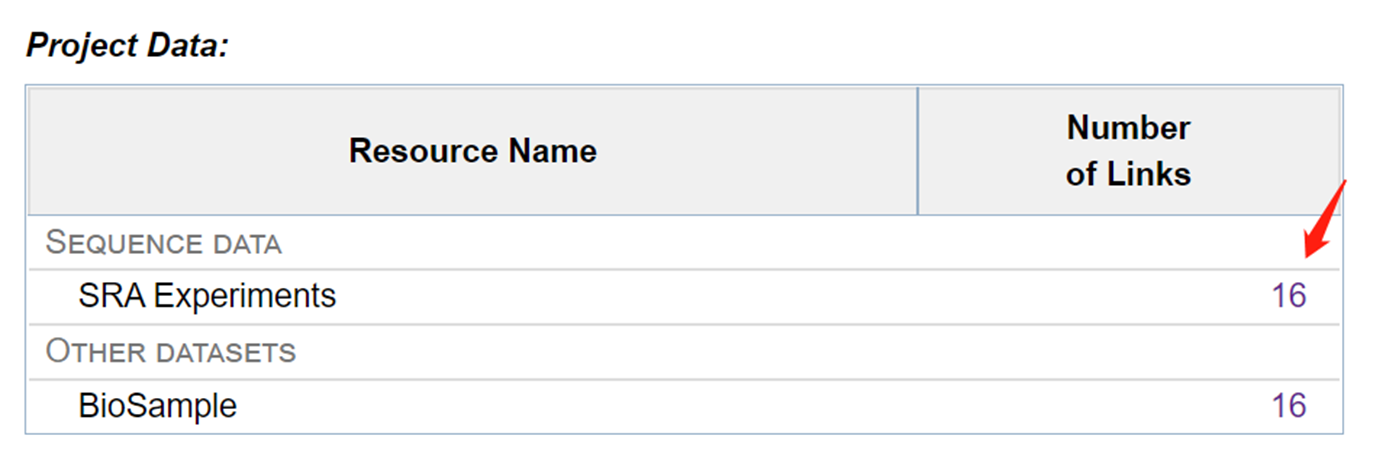
文章中用于分析的样本有16个,下载对应样本的SRA编号即可:
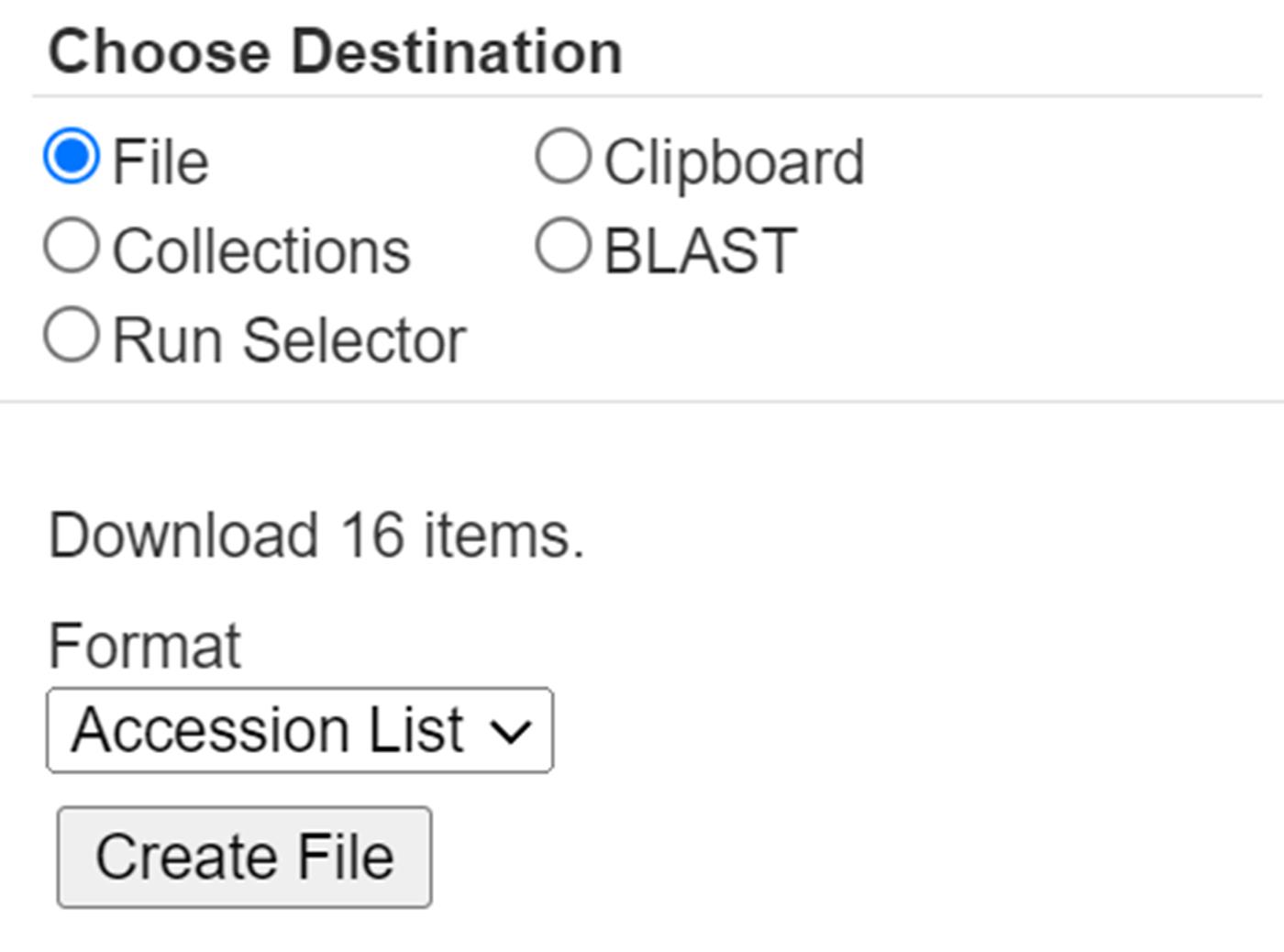
 (2)SRA数据格式转换
(2)SRA数据格式转换
#工作目录:00.raw_data#存放到一个文件夹下mkdir sra_filefor id in `ls -d D*`;do mv ${id}/${id}.sra sra_file;done#格式转换ls sra_file/* > sra_id.txtfor id in `cat sra_id.txt`;do echo "fastq-dump --split-3 --gzip -O ./ ${id}" >> fastq-dump.sh;done# ParaFly运行ParaFly -c fastq-dump.sh -CPU 16 2>fastqdump.err.log# rm -rf sra_file/
arab_ref.fa.fai内容如下,分别代表序列ID、序列长度、序列起始位置(这边计数的时候会把每一条序列的长度也算进来)、每一行有多少个base、每一行有多少个字节。
130427671556061219698289309349096061323459830509615586061418585056748124416061526975502937073036061Mt3669241211324526061Pt1544781215055476061
(3)质量过滤
#工作目录;00.raw_data# mkdir ../01.clean_datacat SraAccList.txt | while read iddo echo "fastp -i ${id}_1.fastq.gz -I ${id}_2.fastq.gz -o ../01.clean_data/${id}_clean_1.fastq.gz -O ../01.clean_data/${id}_clean_2.fastq.gz --cut_tail --n_base_limit 3 --length_required 60 --correction --thread 4 --html ../01.clean_data/${id}.html --json ../01.clean_data/${id}.json" >> fastp.commandlinesdone# ParaFly运行ParaFly -c fastp.commandlines -CPU 16 2>fastp.err.log &
(4)比对
1、bwa比对
# mkdir ../02.bwacat ../00.raw_data/SraAccList.txt | while read id;do echo "bwa mem -t 6 -R '@RG\tID:foo\tPL:illumina\tSM:${id}' ../00.ref/arab_ref.fa ${id}_clean_1.fastq.gz ${id}_clean_2.fastq.gz | samtools view -Sb - > ../02.bwa/${id}.unsorted.bam" >> bwa.commondlines; done# ParaFly运行ParaFly -c bwa.commondlines -CPU 8 2>bwa_mem.err.log &
没有经过排序的BAM文件,第4列还是乱序:
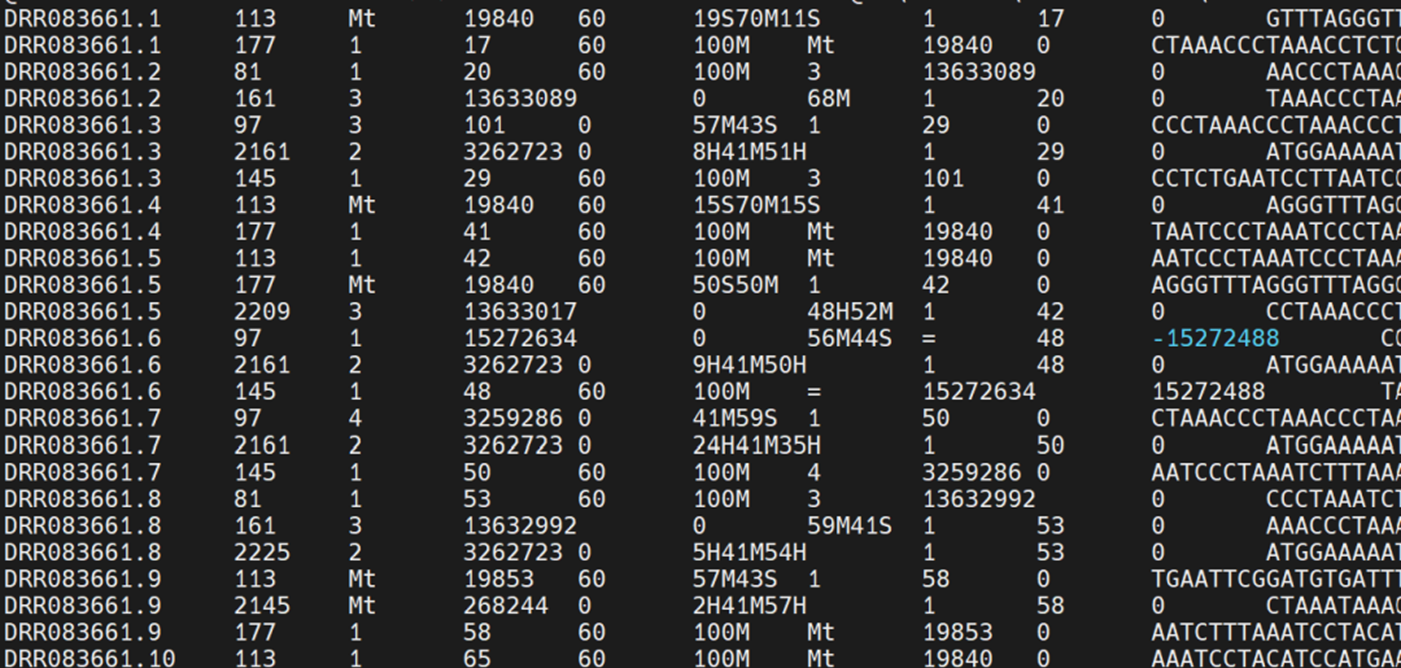
2、samtools排序
ls *.bam > unsorted_bam_id.txtcat unsorted_bam_id.txt | while read iddo name=${id%%.*} echo "samtools sort -@ 6 -m 4G -O bam -o ${name}.sorted.bam ${id}" >> sortbam.commandlinesdone# ParaFly运行ParaFly -c sortbam.commandlines -CPU 16 2>sortbam.err.log &#删除unsorted# ls * | grep "unsorted.bam$" | xargs -i rm -rf {}
sort过后的bam文件:
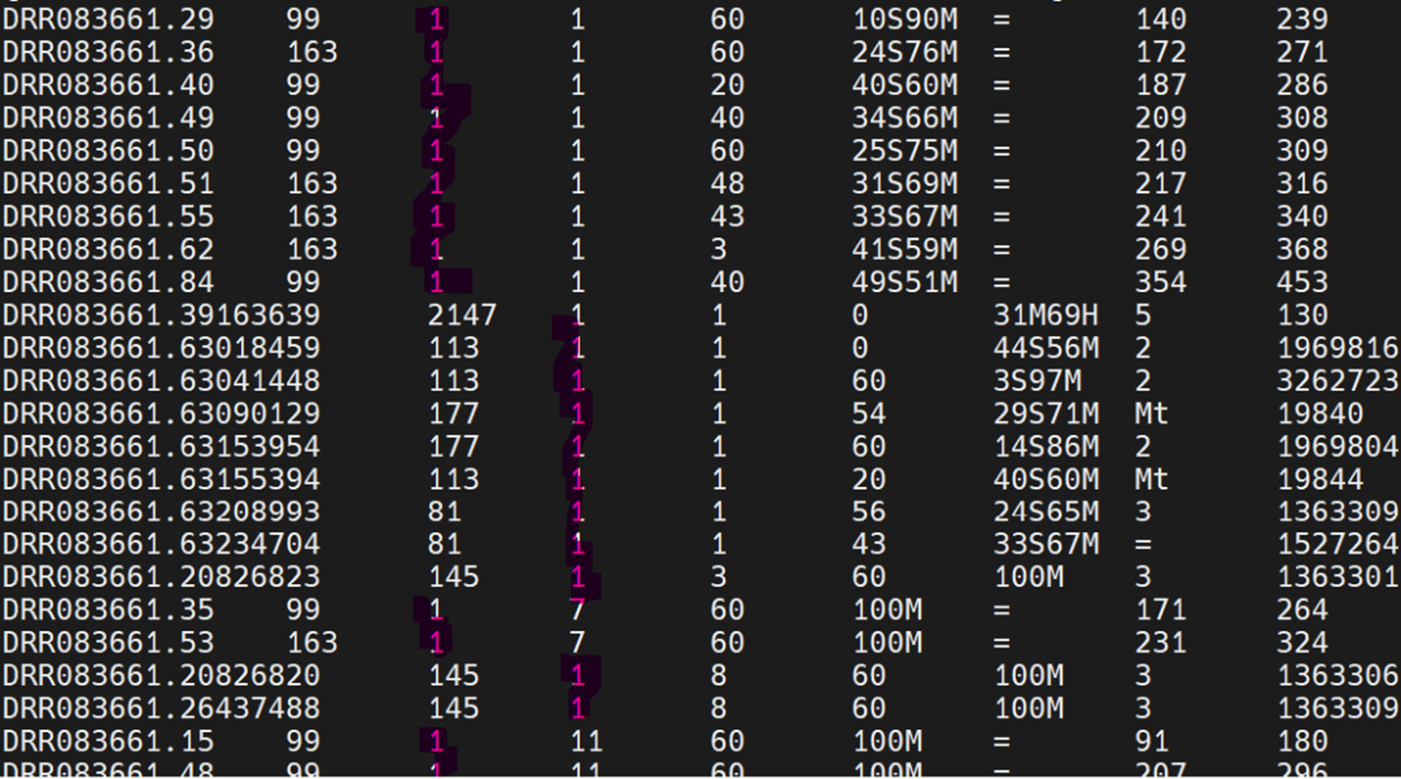
(5)标记重复序列
在NCBI官网查看了每一个样本的建库情况,并不是全部都说明了使用PCR-free kit(甚至没有说明),保险起见还是对PCR duplicates进行标记。
ls *.sorted.bam > sorted_bam_id.txtcat sorted_bam_id.txt | while read iddo name=${id%%.*} echo "java -jar ~/biosoft/picard.jar MarkDuplicates I=${id} O=${name}.markdup.bam M=${name}.markdup.metrc.csv" >> markdup.commandlinesdone# ParaFly运行ParaFly -c markdup.commandlines -CPU 16 2>markdup.err.log &# rm -rf *.sorted.bam#上述这一步骤完成之后,需要对结果文件建立索引,以.bai结尾for id in `ls *.markdup.bam`;do echo "java -jar ~/biosoft/picard.jar BuildBamIndex I=${id}" >> buildbamindex.commondlines;done# ParaFly运行ParaFly -c buildbamindex.commondlines -CPU 16 2>buildbamindex.err.log &
上述建立索引这一步,也可以在运行MarkDuplicates就添加,即[--CREATE_INDEX]
(6)SNP calling
#构建参考序列dict文件java -jar ~/biosoft/picard.jar CreateSequenceDictionary R=../00.ref/arab_ref.fa O=../00.ref/arab_ref.dict# 1、HaplotypeCaller# mkdir 03.snpcalling && cd 03.snpcallingls ../02.bwa/*.bam | xargs -i basename {} > bam_id.txtcat bam_id.txt | while read iddo name=${id%%.*} echo "gatk --java-options \"-Xmx15g\" HaplotypeCaller -R ../00.ref/arab_ref.fa -I ../02.bwa/$id -O ${name}.g.vcf.gz -ERC GVCF" >> haplotypecaller.commondlinesdoneParaFly -c haplotypecaller.commondlines -CPU 16 2>haplotypecaller.err.log &
这些gvcf文件的大小差异有点大,基于该实验设计似乎又蛮合理,不同程度的辐射对突变位点数量的影响肯定是不一样的,但是这还是只是GATK分析的第一步,所以按要求过滤完了之后再下定论也不迟。
# 2、GenomicsDBImportls *.g.vcf.gz > gvcf_id.txtcut -d "." -f 1 gvcf_id.txt > sample_id.txtpaste -d "\t" sample_id.txt gvcf_id.txt > sample.mapmy_database=arab_16_DB #创建GenomicsDatabase的文件夹名称samplemap=sample.map #使用tab分隔符,第一列为sampleID,第二列为对应gvcf IDinterval_list=interval.list #每一行一个区域编号gatk --java-options "-Xmx4g -Xms4g" \GenomicsDBImport \--genomicsdb-workspace-path $my_database \--sample-name-map $samplemap \-L $interval_list 2>genomicsDB_build.err.log &
上述这些步骤还不需要纠结很多分析参数,到了GATK硬过滤的时候,就需要考虑了。
参考资料
[1]https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels-
[2]http://www.htslib.org/doc/faidx.html
转载自《生信菜鸟团》,如有侵权,请联系删除。

