单细胞降维聚类分群大家都很熟悉了,通常是基于R语言的seurat操作和基于Python的Scanpy,但是我们也提到过一下小众产品,比如:单细胞降维聚类分群的另外一个工具选择Pagoda2,如果是单个单细胞转录组样品,那么选择哪一个流程其实大同小异,而且我们也强调大家需要熟练掌握5个R包,比如: scater,monocle,Seurat,scran,M3Drop,总之多多益善啦。
但是现在基本上大家的单细胞转录组项目不太可能是单个样品啦,所以一定会触及到多个样品整合的问题,整合是为了尽可能的去除批次等不需要的差异但是尽可能的保留生物学差异,是一个两难问题,所以关于它的算法基本上都是发表在CNS及其子刊。如下所示:
MarkdownLanguageLibraryRefCCARSeuratCellMNNR/PythonScater/ScanpyNat. Biotech.ConosRconosNat. MethodsScanoramaPythonscanoramaNat. Biotech.
实际操作种,因为内存等计算机资源限制,我们并不会选择Seurat体系的CCA方法,而是harmony替代啦。但是如果你选择:单细胞降维聚类分群的另外一个工具选择Pagoda2,其实也有一个配套的单细胞数据集整合的算法选择conos,让我们来一起看看吧。
它的GitHub官网是:https://github.com/kharchenkolab/conos,感兴趣的可以读一下:
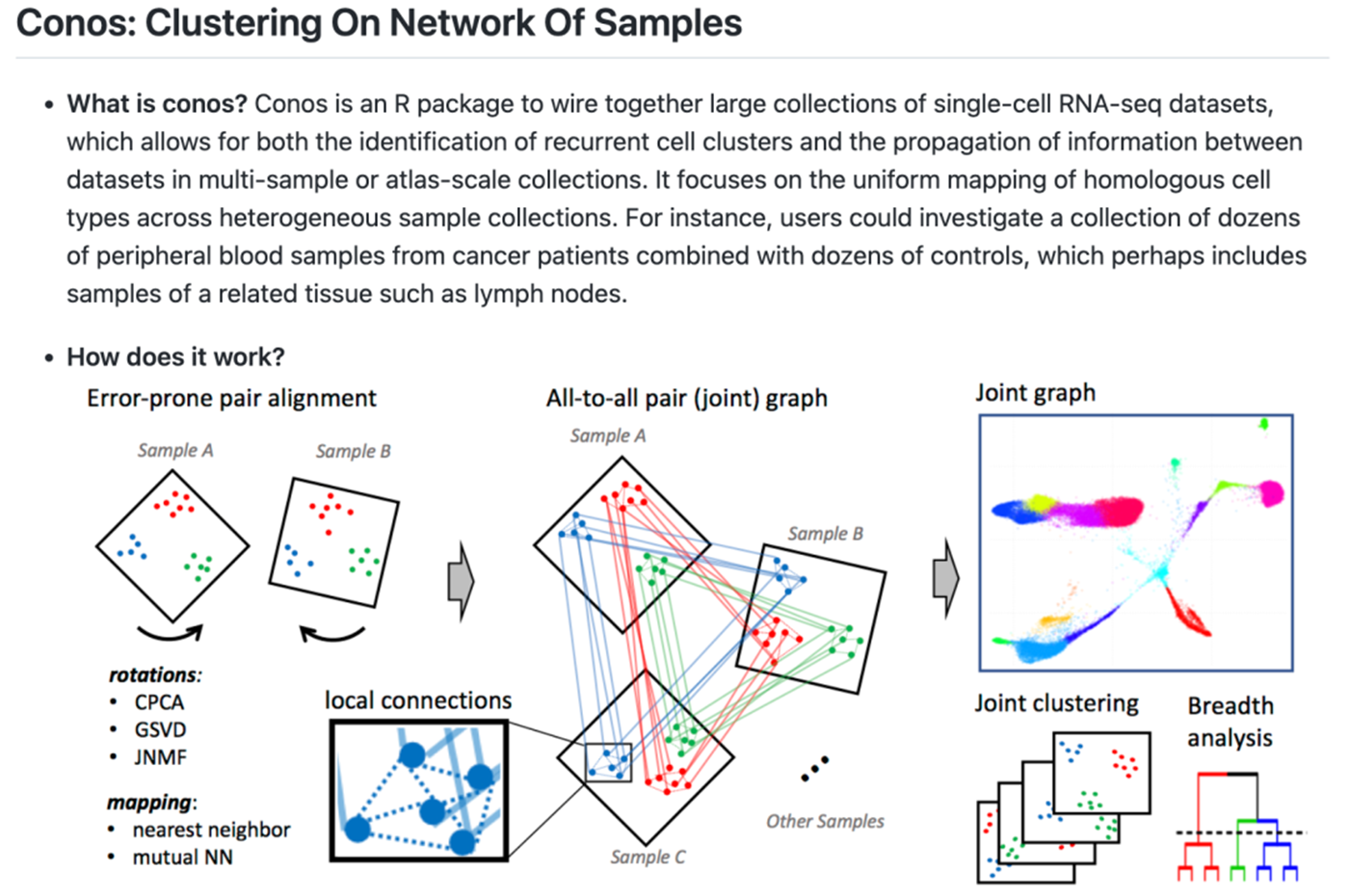
其主要的步骤和函数如下所示:
# Construct Conos object, where pl is a list of pagoda2 objects con <- Conos$new(pl)# Build joint graphcon$buildGraph()# Find communitiescon$findCommunities()# Generate embeddingcon$embedGraph()# Plot joint graphcon$plotGraph()# Plot panel with joint clustering resultscon$plotPanel()
下面让我们通过示例数据,以及实战数据来说明这个包的用法吧。
首先安装和加载常见的包:
# 1.加载R包####rm(list = ls())library(Seurat)library(tidyverse)library(ggsci)library(pagoda2) library(Matrix) library(conos)library(dplyr)library(entropy)# install.packages('conosPanel', repos='https://kharchenkolab.github.io/drat/', type='source')# install.packages("conos")
然后对测试数据集构建Conos对象
# 2.加载数据###### 2.1测试数据----library(conosPanel)panel <- conosPanel::panel # panel是一个List,包含4个单细胞样本的表达量稀疏矩阵#而且都是3000个细胞,3万多个基因lapply(panel, dim)###用Seurat对4个单细胞样品都进行预处理library(Seurat)panel.preprocessed.seurat <- lapply(panel, basicSeuratProc)## 2.2构建Conos对象 ----con <- Conos$new(panel.preprocessed.seurat, n.cores=1)
我们的4个单细胞表达量矩阵,经过了basicSeuratProc的处理,其实就是针对每个矩阵都独立的降维聚类分群啦,感兴趣的可以去看basicSeuratProc的源代码。
最后就可以进行降维聚类分群和整合
space='PCA' #可以选择PCA, CPCA,CCAcon$buildGraph(k=30, k.self=5, space=space, # PCA, CPCA,CCA ncomps=30, n.odgenes=2000, matching.method='mNN', metric='angular', score.component.variance=TRUE, verbose=TRUE)plotComponentVariance(con, space=space) ## 2.3 leiden.community法分群 ----resolution = 1 #可以适当修改分群con$findCommunities(method=leiden.community, resolution=resolution) #相当于Seurat包中的FindClusters函数con$plotPanel(font.size=4) #绘图
可以看到,这4个样品都有各自的降维坐标体系,但是它们的聚类分群是一致的,如果分开可视化,就是con$plotPanel(font.size=4)函数,结果如下所示:
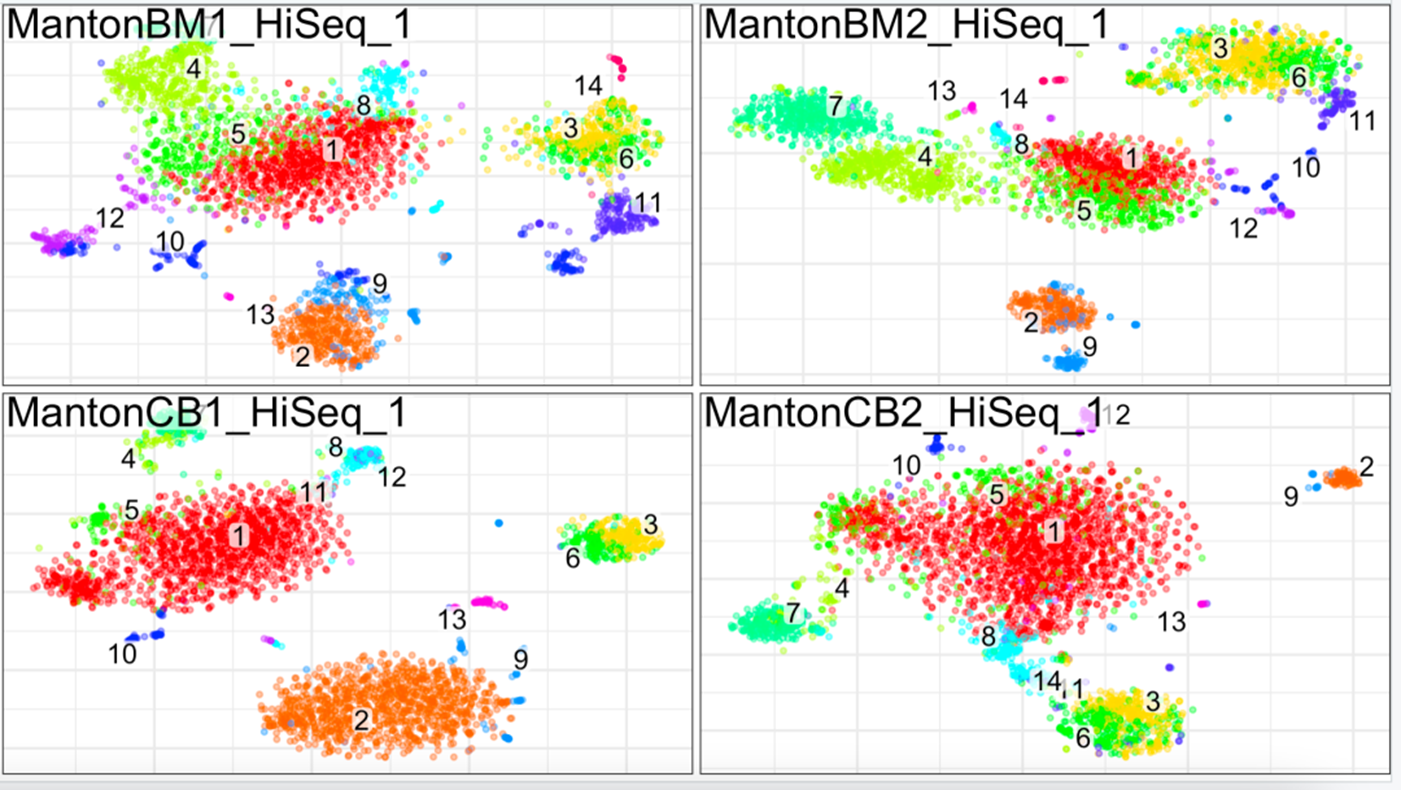
分开可视化
如果合并可视化,代码如下所示:
table(con$clusters$leiden$groups) con$embedGraph(method='largeVis') ## 2.4整合后后的效果展示----library(patchwork)con$plotGraph(clustering='leiden') + con$plotGraph(color.by='sample', mark.groups=FALSE, alpha=0.1, show.legend=TRUE)#可视化每个cluster不同样本的占比plotClusterBarplots(con, legend.height = 0.1)
可以看到4个样品整合后的分群在各个样品都有分布,说明确实整合在了一起:
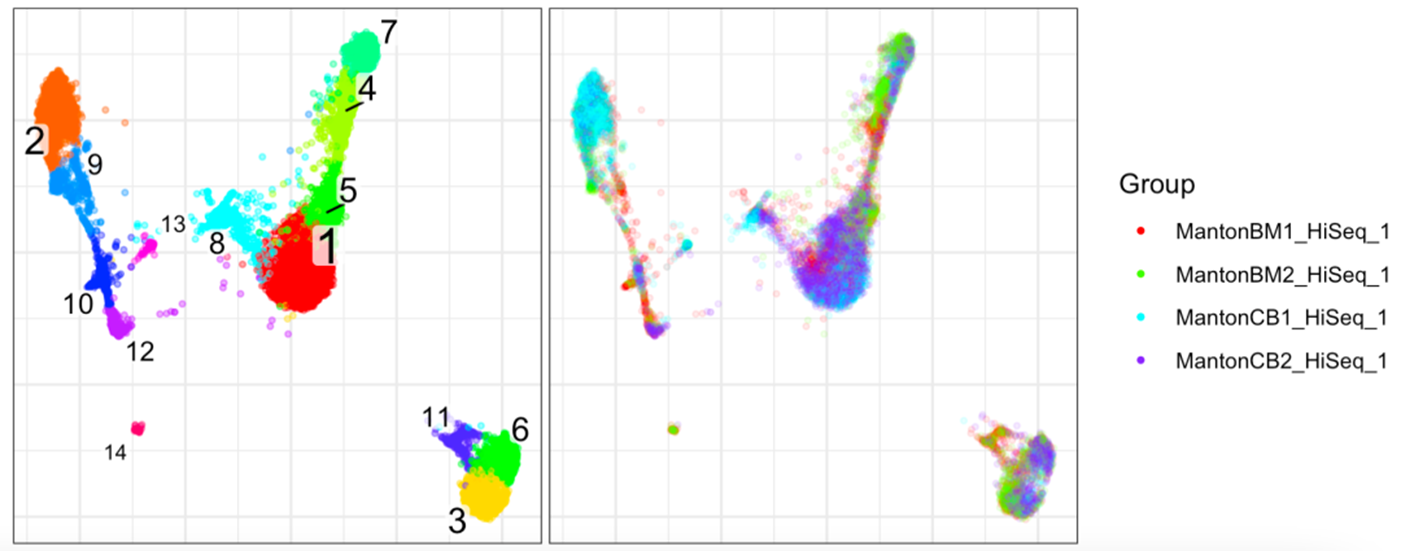
确实整合在了一起
在PBMC实例数据演示conos的整合
前面的包的安装和加载是一样的,这个时候不选择示例数据,而是读取pbmc3k和5k数据集:
## 2.1读取pbmc3k和5k数据集----library(conosPanel)options(stringsAsFactors = F)load('pbmc3k.Rdata')pbmc_3k=pbmcpbmc_5k=readRDS('pbmc_5k_v3.rds') library(Seurat)panel.preprocessed.seurat <- list( pbmc_3k=pbmc_3k,pbmc_5k=pbmc_5k)
这个时候的读取pbmc3k和5k数据集,需要的两个文件在我自己的电脑,不过如果你看完了以前的单细胞系列教程,应该是很容易自己去制作它。
同样的构建Conos对象,并且整合,降维聚类分群,代码如下所示:
## 2.2构建Conos对象 ----con <- Conos$new(panel.preprocessed.seurat, n.cores=1) space='PCA' #可以选择PCA, CPCA,CCAcon$buildGraph(k=30, k.self=5, space=space, # PCA, CPCA,CCA ncomps=30, n.odgenes=2000, matching.method='mNN', metric='angular', score.component.variance=TRUE, verbose=TRUE)plotComponentVariance(con, space=space) ## 2.3聚类分群 ----resolution = 1 #可以适当修改分群con$findCommunities(method=leiden.community, resolution=resolution) #相当于Seurat包中的FindClusters函数con$plotPanel(font.size=4) #绘图table(con$clusters$leiden$groups) con$embedGraph(method='largeVis')con$plotGraph(clustering='leiden')## 2.4整合后后的效果展示----library(patchwork)con$plotGraph(clustering='leiden') + con$plotGraph(color.by='sample', mark.groups=FALSE, alpha=0.1, show.legend=TRUE)
我们也是直接看效果,整合的还不错;
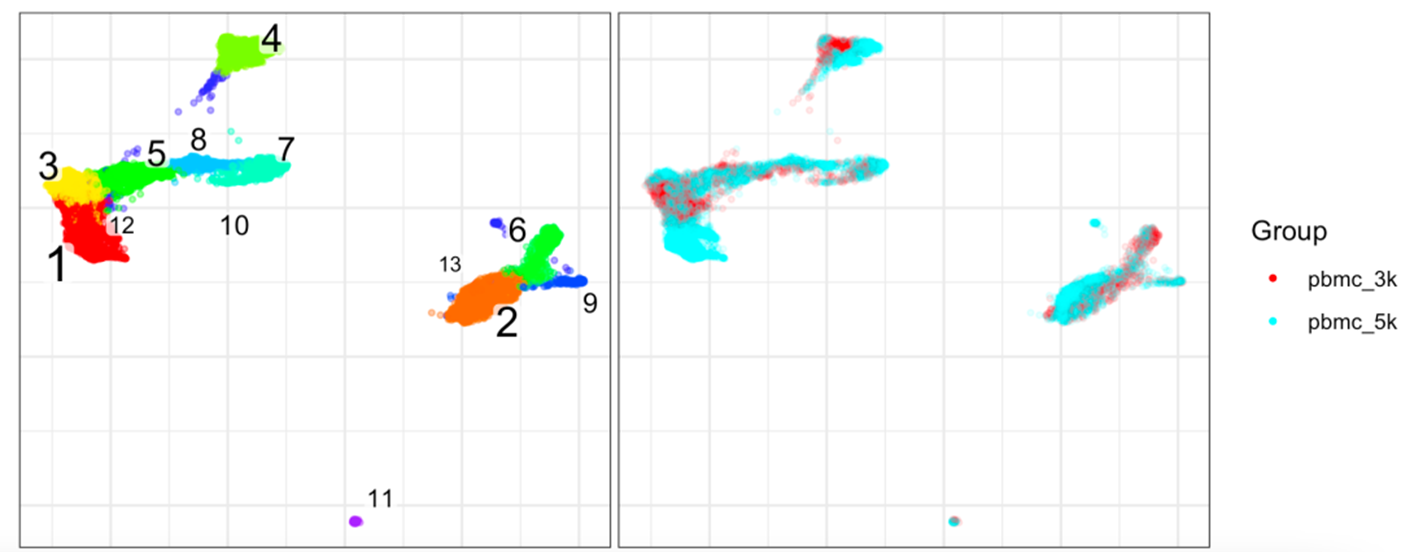
而且因为我们对PBMC比较熟悉,也可以拿常见的基因去可视化:
#可视化基因表达情况,相当于FeaturePlot# T Cells (CD3D, CD3E, CD8A), # B cells (CD19, CD79A, MS4A1 [CD20]), # Plasma cells (IGHG1, MZB1, SDC1, CD79A), # Monocytes and macrophages (CD68, CD163, CD14),# NK Cells (FGFBP2, FCG3RA, CX3CR1), library(patchwork)gene_to_check= c('PTPRC', 'CD3D', 'CD3E','IL7R','CD4','CD8A','CD19', 'CD79A', 'MS4A1')pl = lapply(gene_to_check, function(gene){ con$plotGraph(gene = gene,title=paste0(gene,' expression'), alpha=0.5)})wrap_plots(pl, byrow = T, nrow = 3)
可以比较容易的看到各种免疫细胞啦:
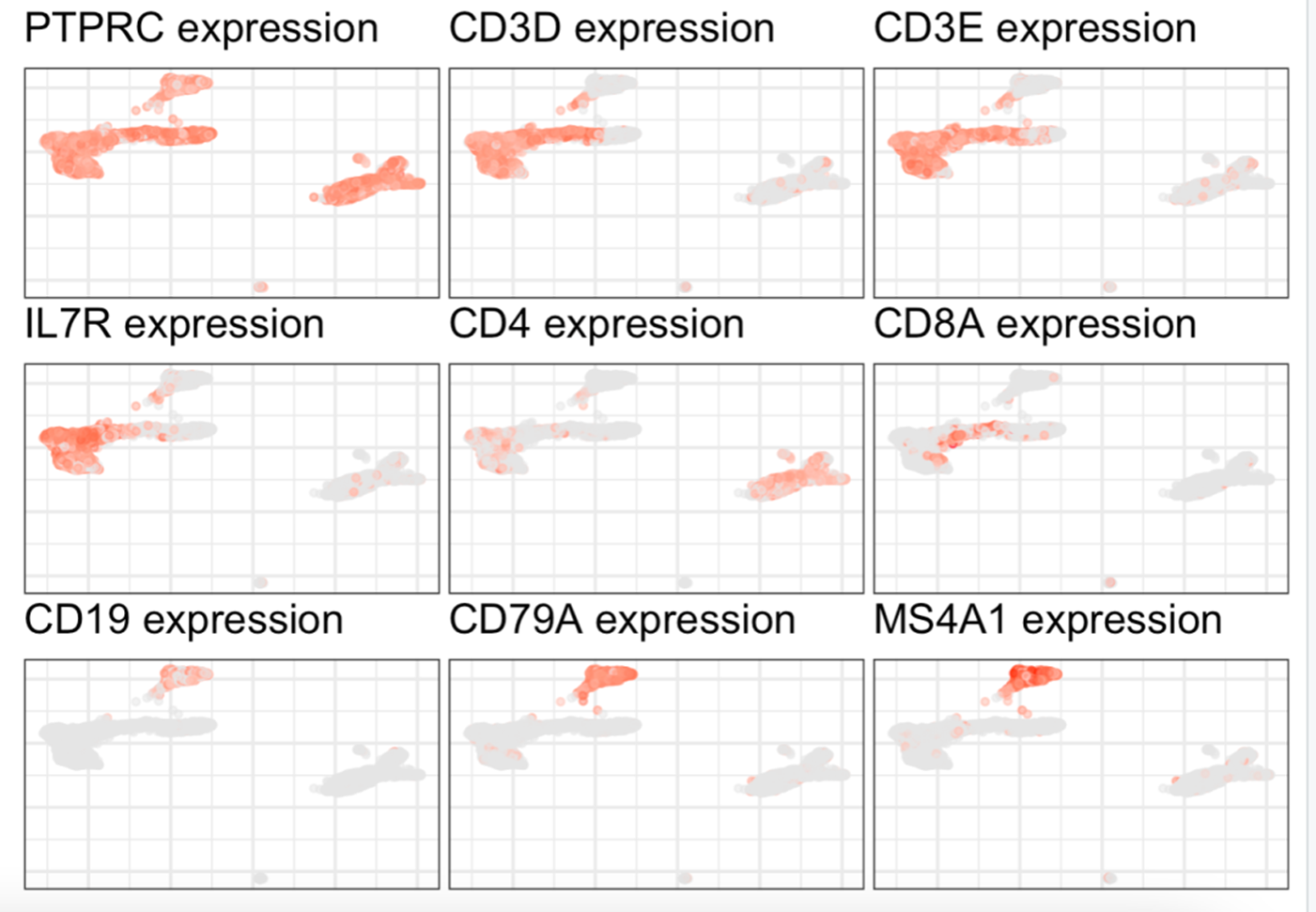
各种免疫细胞的标记基因
值得注意的是这个算法的引用非常差,目前为止(2022-04-03)还不到100的引用,跟其它单细胞算法比起来都是数量级的差异。
转载自《生信技能树》,如有侵权,请联系删除。

