Oncogenic lncRNA downregulates cancer cell antigen presentation and intrinsic tumor suppression不过不需要看文章,大家只需要做差异分析即可,这个时候需要注意的是,作者提供的是RPKM值表达矩阵!
1. 下载数据GSE113143并加载数据
a=read.table('GSE113143_Normal_Tumor_Expression.tab.gz',sep='\t',quote = "",fill = T, comment.char = "!",header = T) #提取表达矩阵rownames(a)=a[,1]a <- a[,-1]
1. 将FPKM转换为TPM
Q:为什么将FPKM转换为TPM?A:只有转换成TPM才勉强可以用limma做差异分析;而DESeq2和edgeR是对count数据进行差异分析
expMatrix <- afpkmToTpm <- function(fpkm){ exp(log(fpkm) - log(sum(fpkm)) + log(1e6))}tpms <- apply(expMatrix,2,fpkmToTpm)tpms[1:3,]colSums(tpms)#输出结果:> tpms[1:3,] N1 N2 N3 T1 T2 T30610005C13Rik 0.232 0.1715 0.00 0.00 0.00 0.000610007P14Rik 48.391 39.2632 46.04 50.04 59.05 67.290610009B22Rik 47.491 58.5954 54.27 49.79 53.13 58.00> colSums(tpms) N1 N2 N3 T1 T2 T3 1e+06 1e+06 1e+06 1e+06 1e+06 1e+06
1. 差异分析
group_list=c(rep('Normal',3),rep('Tumor',3))##强制限定顺序group_list <- factor(group_list,levels = c("Normal","Tumor"),ordered = F)#表达矩阵数据校正exprSet <- tpmsboxplot(exprSet,outline=FALSE, notch=T,col=group_list, las=2)library(limma) exprSet=normalizeBetweenArrays(exprSet)boxplot(exprSet,outline=FALSE, notch=T,col=group_list, las=2)#判断数据是否需要转换exprSet <- log2(exprSet+1)#差异分析:dat <- exprSetdesign=model.matrix(~factor( group_list ))fit=lmFit(dat,design)fit=eBayes(fit)options(digits = 4)topTable(fit,coef=2,adjust='BH')bp=function(g){ library(ggpubr) df=data.frame(gene=g,stage=group_list) p <- ggboxplot(df, x = "stage", y = "gene", color = "stage", palette = "jco", add = "jitter") # Add p-value p + stat_compare_means()}deg=topTable(fit,coef=2,adjust='BH',number = Inf)head(deg) #save(deg,file = 'deg.Rdata')
这里面重点就是:RPKM矩阵可以转为TPM后,再使用limma进行差异分析哦!
1. 做完差异分析
##不同的阈值,筛选到的差异基因数量就不一样,后面的超几何分布检验结果就大相径庭。if(T){ logFC_t=1.5 deg$g=ifelse(deg$P.Value>0.05,'stable', ifelse( deg$logFC > logFC_t,'UP', ifelse( deg$logFC < -logFC_t,'DOWN','stable') ) ) table(deg$g) head(deg) deg$symbol=rownames(deg) library(ggplot2) library(clusterProfiler) library(org.Mm.eg.db) df <- bitr(unique(deg$symbol), fromType = "SYMBOL", toType = c( "ENTREZID"), OrgDb = org.Mm.eg.db) head(df) DEG=deg head(DEG) DEG=merge(DEG,df,by.y='SYMBOL',by.x='symbol') head(DEG) save(DEG,file = 'anno_DEG.Rdata') gene_up= DEG[DEG$g == 'UP','ENTREZID'] gene_down=DEG[DEG$g == 'DOWN','ENTREZID'] }
1. 最简单的超几何分布检验
#最简单的超几何分布检验###这里就拿KEGG数据库举例吧,拿自己判定好的上调基因集进行超几何分布检验,如下if(T){ gene_down gene_up enrichKK <- enrichKEGG(gene = gene_up, organism = 'mmu', #universe = gene_all, pvalueCutoff = 0.05, qvalueCutoff =0.05) head(enrichKK)[,1:6] browseKEGG(enrichKK, 'hsa04512') dotplot(enrichKK) ggsave("enrichKK.png") enrichKK=DOSE::setReadable(enrichKK, OrgDb='org.Mm.eg.db',keyType='ENTREZID') enrichKK }##最基础的条形图和点图#条带图barplot(enrichKK,showCategory=20)#气泡图dotplot(enrichKK)
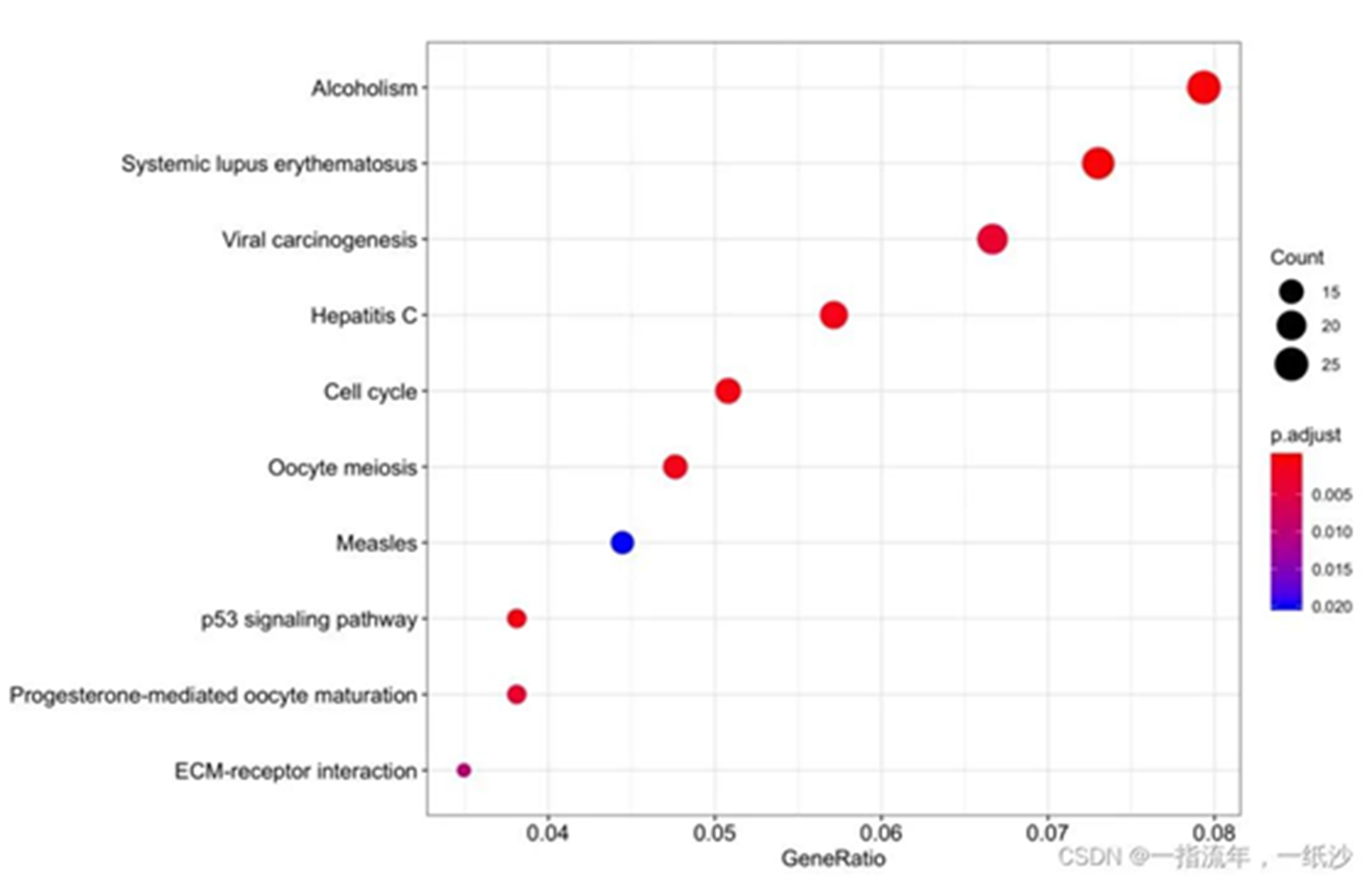 通路与基因之间的关系可视化
通路与基因之间的关系可视化
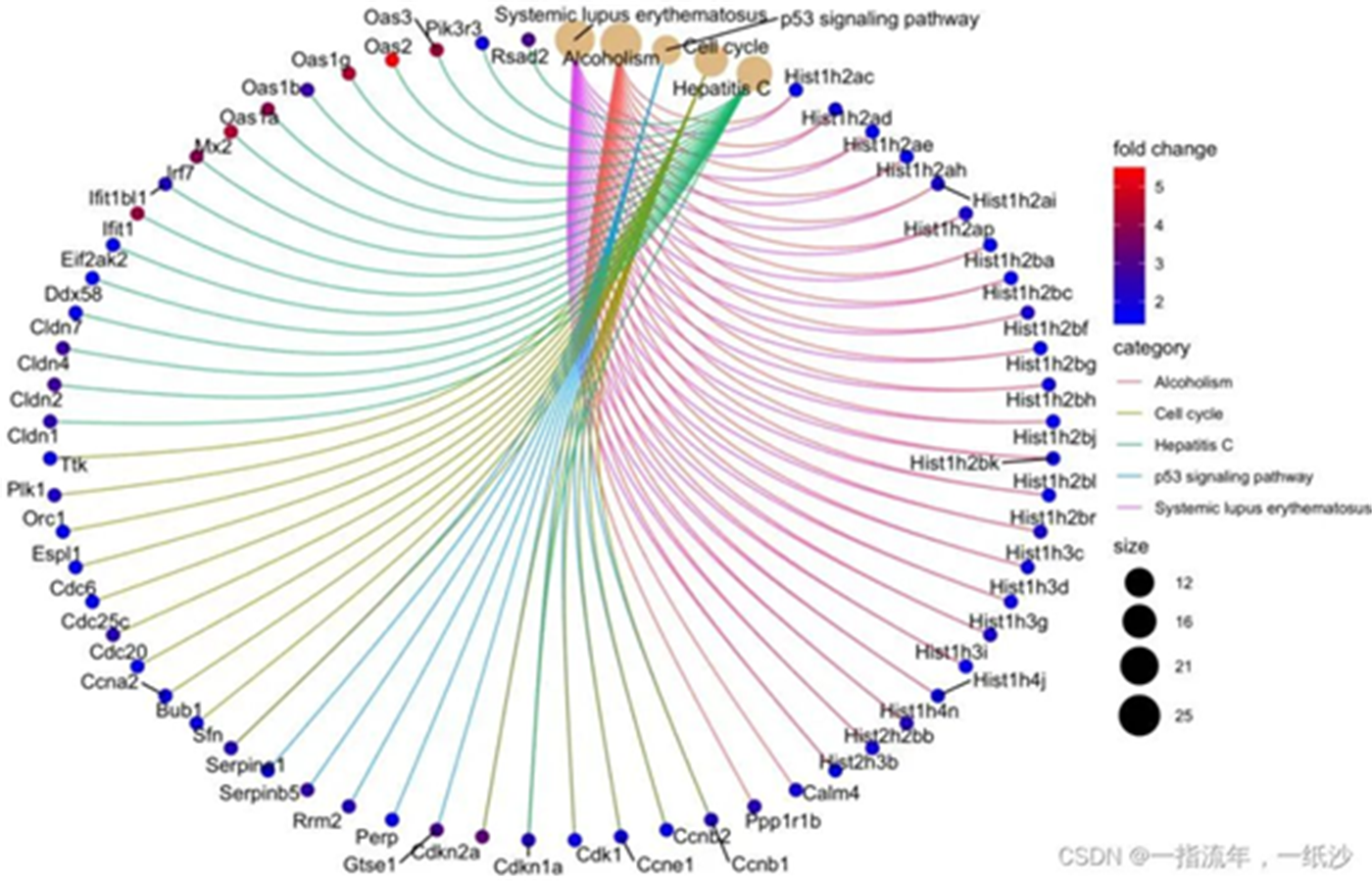
#通路与上调基因之间的关系可视化###制作genlist三部曲:## 1.获取基因logFC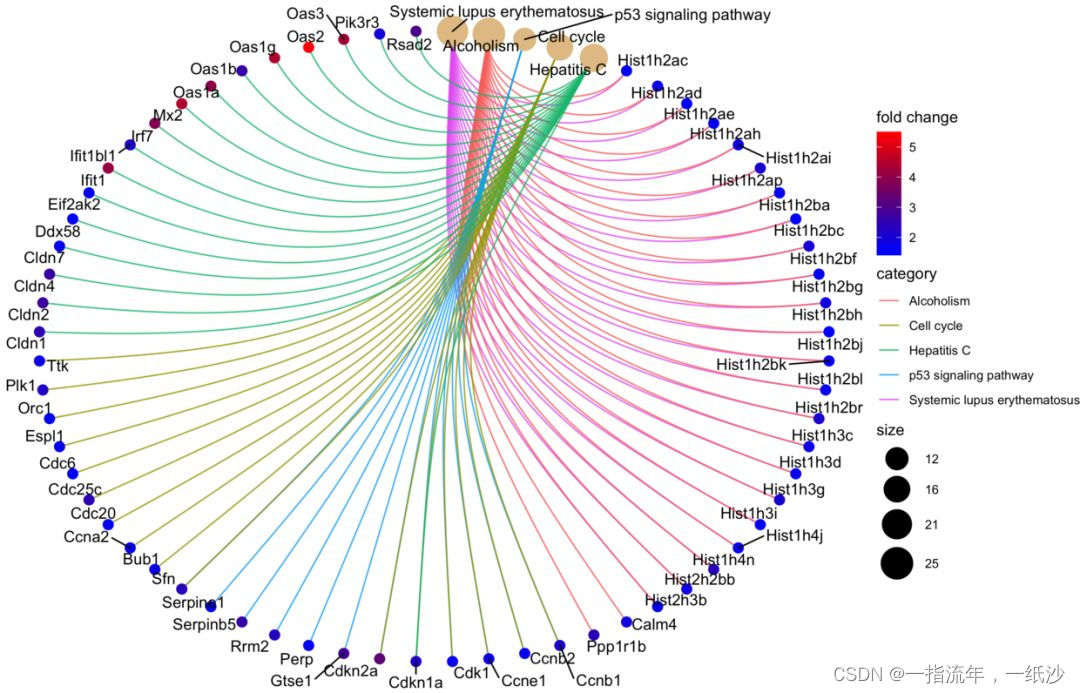DEG_up <- DEG[DEG$g == 'UP',]geneList <- DEG_up$logFC## 2.命名names(geneList) = DEG_up$ENTREZID## 3.排序很重要geneList = sort(geneList, decreasing = TRUE)head(geneList)cnetplot(enrichKK, categorySize="pvalue", foldChange=geneList,colorEdge = TRUE)cnetplot(enrichKK, foldChange=geneList, circular = TRUE, colorEdge = TRUE)ggsave("enrichKK_cnetplot.png")
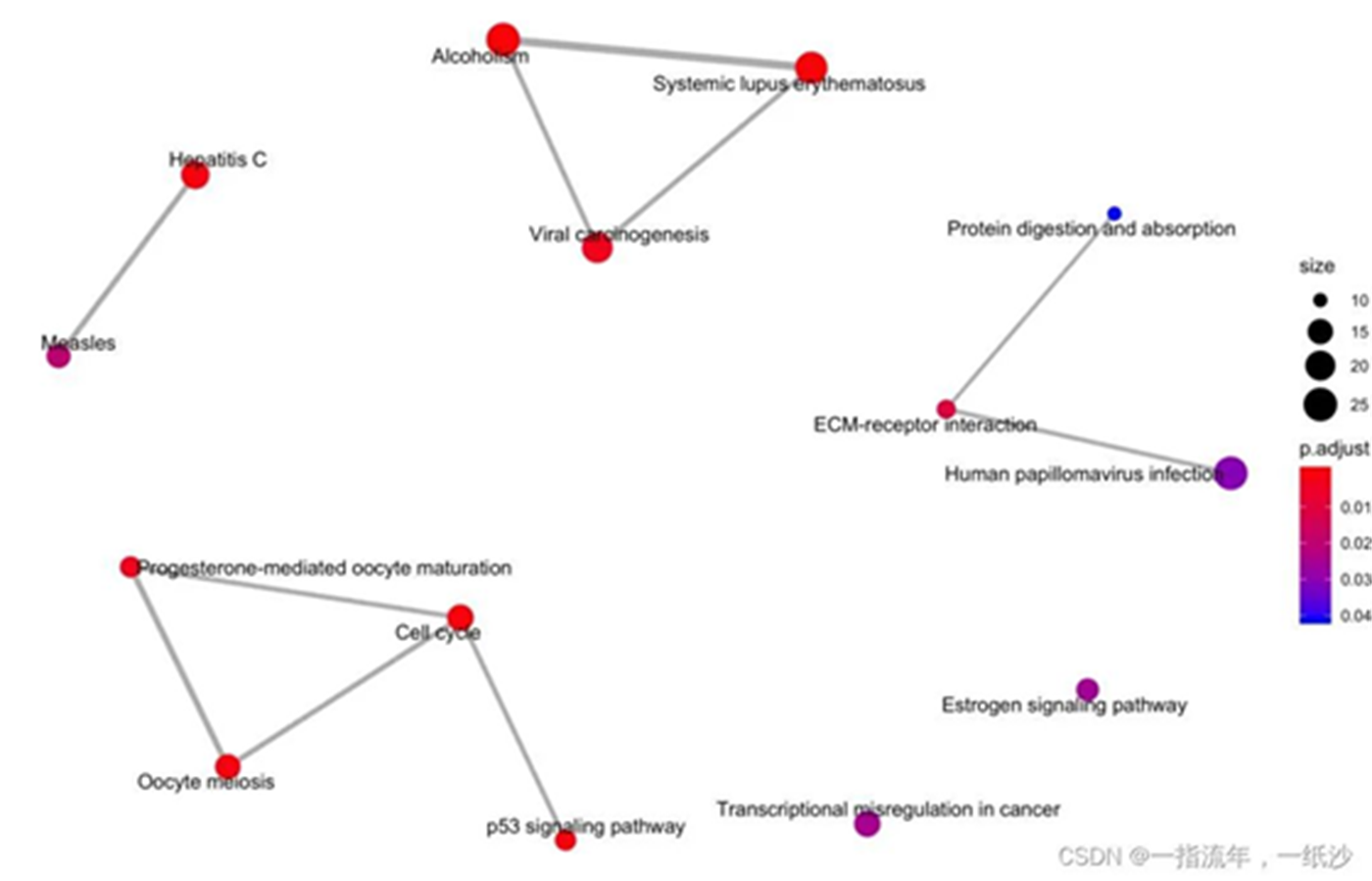
 通路与通路之间的连接展示
通路与通路之间的连接展示
#通路与通路之间的连接展示emapplot(enrichKK)ggsave("enrichKK_emapplot.png")
 热图展现通路与基因之间的关系
热图展现通路与基因之间的关系
#热图展现通路与基因之间的关系heatplot(enrichKK)ggsave("enrichKK_heatplot.png")
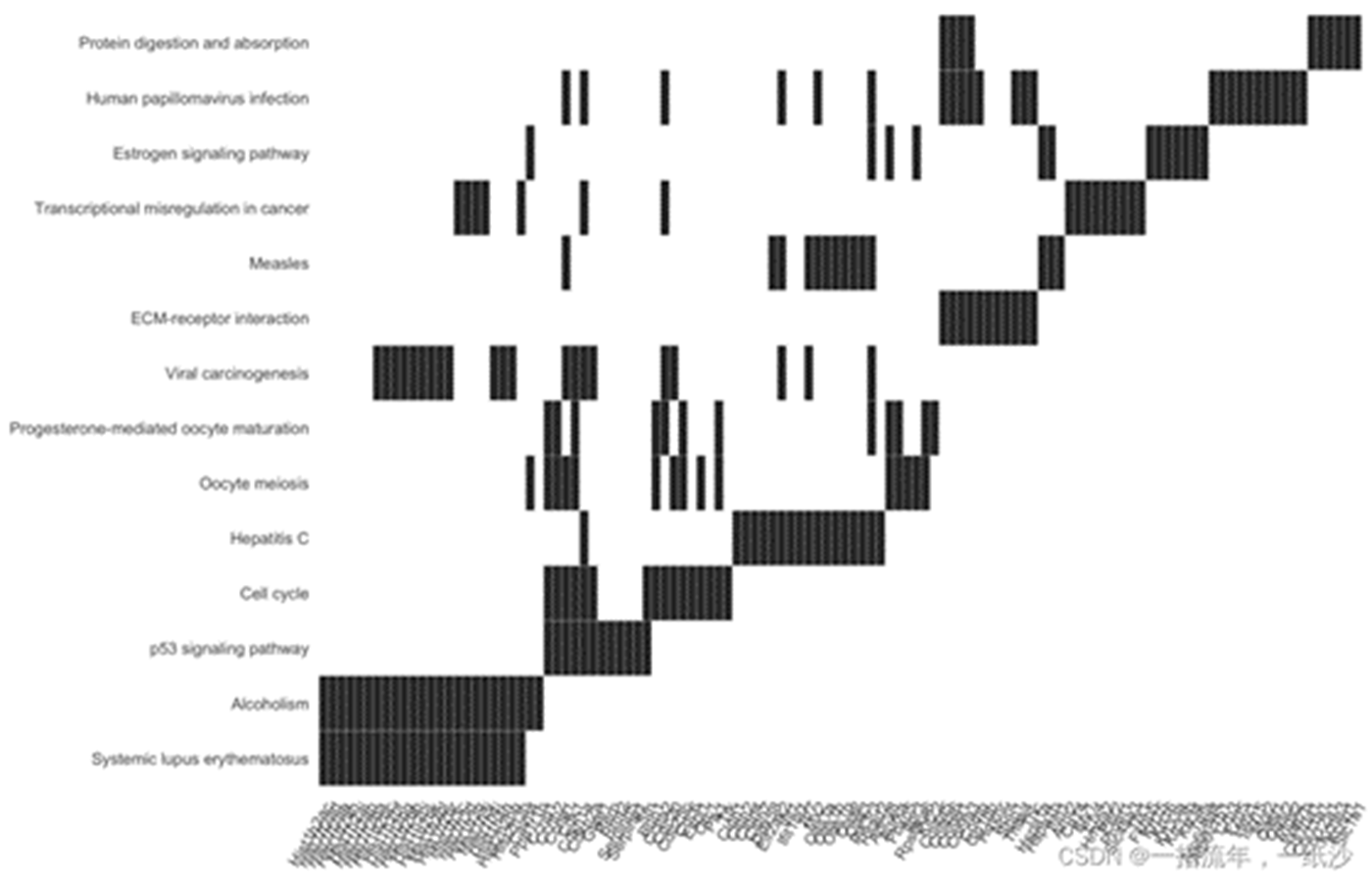 如果你是做GO数据库呢,其实还有一个goplot可以试试看
如果你是做GO数据库呢,其实还有一个goplot可以试试看
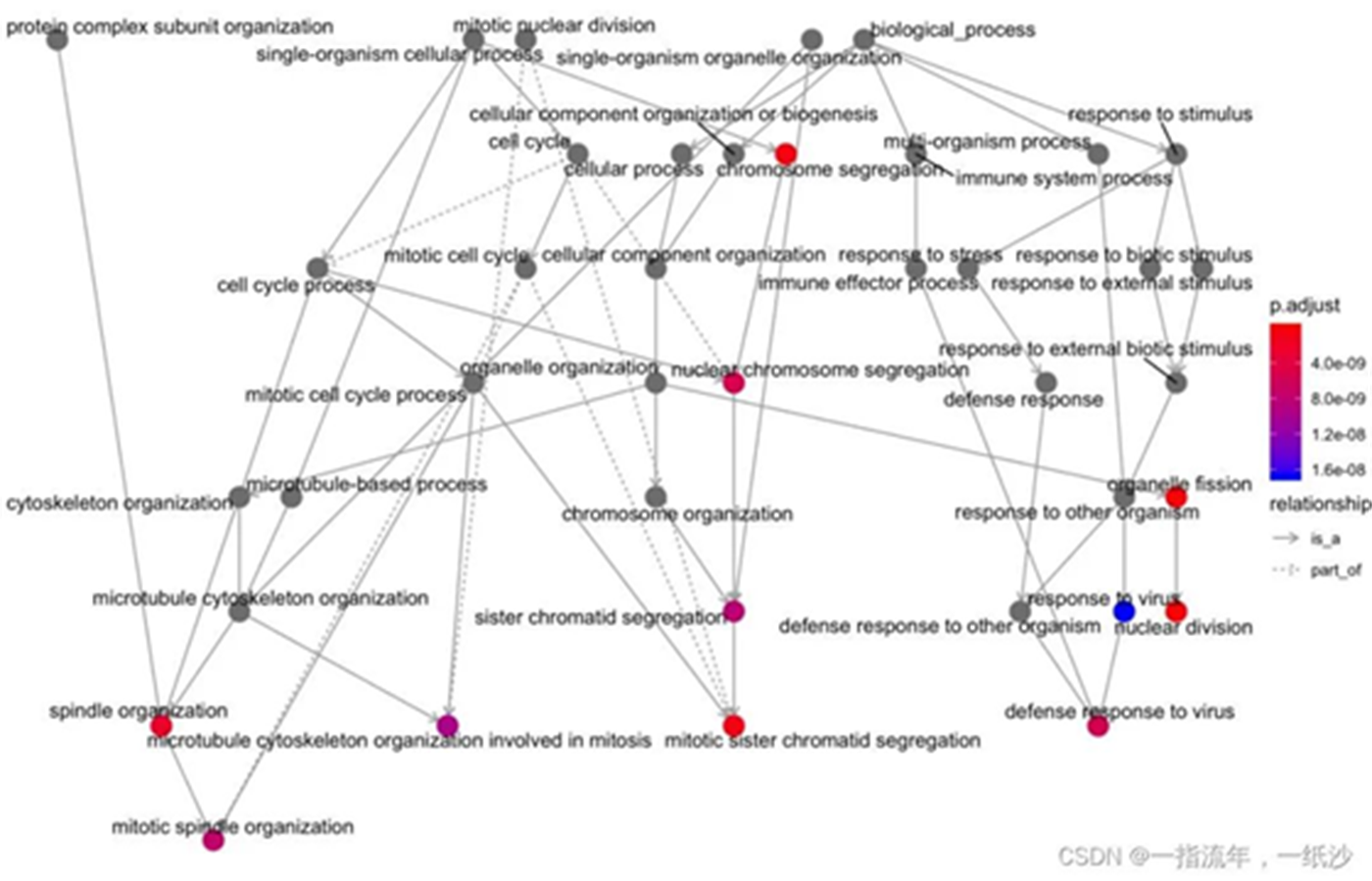
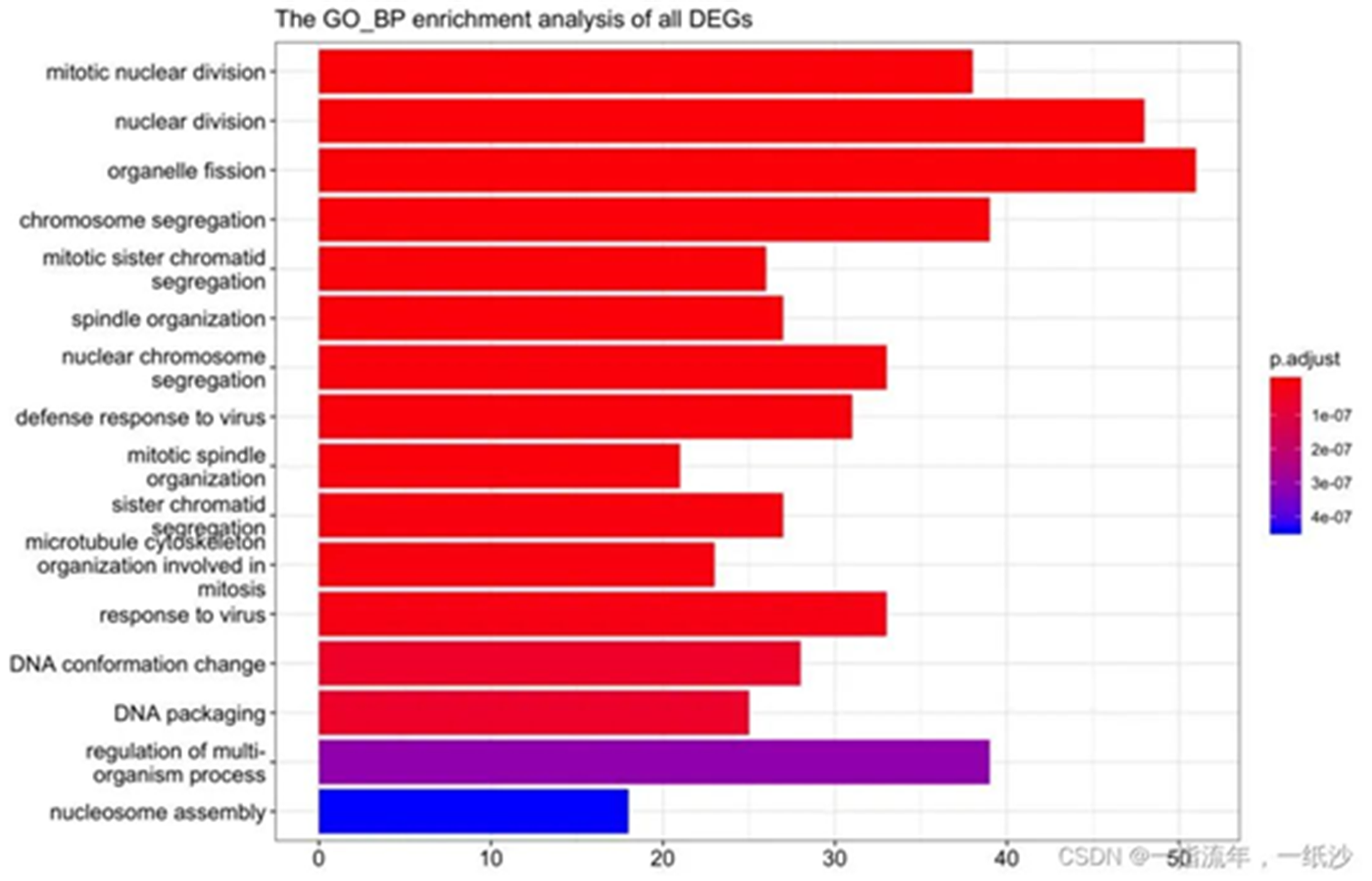
#如果你是做GO数据库呢,其实还有一个goplot可以试试看ego_bp_up<-enrichGO(gene = DEG_up$ENTREZID, OrgDb = org.Mm.eg.db, keyType = 'ENTREZID', ont = "BP", pAdjustMethod = "BH", pvalueCutoff = 0.01,#0.01 qvalueCutoff = 0.05)goplot(ego_up)ggsave("ego_bp_up_goplot.png")head(ego)library(stringr)barplot(ego_bp_up,showCategory = 16,title="The GO_BP enrichment analysis of all DEGs ")+ scale_size(range=c(2, 12))+ scale_x_discrete(labels=function(ego_bp) str_wrap(ego_bp,width = 25))ggsave("ego_bp_up_barplot.png")
转载自《生信技能树》,如有侵权,请联系删除。

