本文由医信融合团队成员“张皓旻”撰写,已同步至微信公众号“医信融合创新沙龙”与“表观精准治疗”,更多精彩内容欢迎关注!


获得转录组数据(.fastq 文件)后的第一步就是对原始数据的质量评估。
目的
质量评估的目的是全面查看原始数据的质量,内容包括碱基质量评估、GC 含量检验、N 碱基数量评估、TCGA 碱基分布、k-mer 数量检验等。
方法
检验 fastq 文件质量的软件有很多,例如 FastQC、fastp、multiQC 等。本文主要介绍应用最多的 FastQC。
FastQC 是一款基于 Java 的软件,须在 linux 环境下使用命令行运行,它可以快速多线程地对测序数据进行质量评估(Quality Control),其官网地址为:Babraham Bioinformatics。
安装
FastQC 可以使用 conda 进行安装。在 linux 环境下运行命令conda install -c bioconda fastqc=0.11.9即可,运行结果如下图。
运行命令fastqc -h可检验其是否成功安装,运行结果如下图。

运行
#运行命令的基本格式
# fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
#主要是包括前面的各种选项和最后面的可以加入N个文件
# -o --outdir FastQC生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的
# --extract生成的报告默认会打包成1个压缩文件,使用这个参数是让程序不打包
# -t --threads选择程序运行的线程数,每个线程会占用250MB内存,越多越快咯
# -c --contaminants污染物选项,输入的是一个文件,格式是Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到
# -a --adapters也是输入一个文件,文件的格式Name [Tab] Sequence,储存的是测序的adpater序列信息,如果不输入,目前版本的FastQC就按照通用引物来评估序列时候有adapter的残留
# -q --quiet安静运行模式,一般不选这个选项的时候,程序会实时报告运行的状况。
使用fastqc -o #输出结果全路径#数据存储全路径/*reads_R1.fq命令运行
对我们在RNA-Seq 数据分析准备—SRA 数据下载及整理中下载并整理好的数据(GSE176393)进行质量评估
运行后可获得如下结果。
结果解读
看懂质量评估的结果是非常重要的!!!
Basic Statistics基本信息
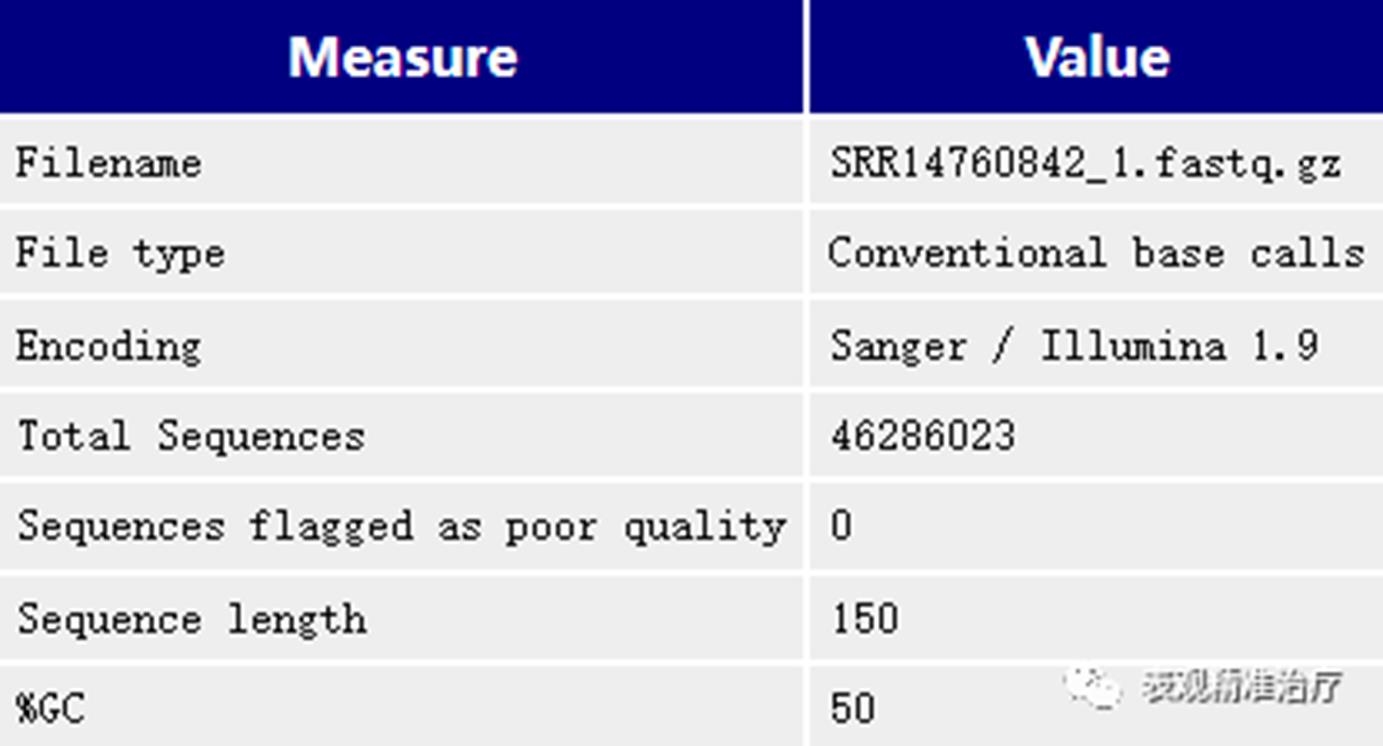
报告第一部分是对质量检测结果的基本信息统计,如上图所示。其中包括:
• Filename:检测的 fastq 文件名称;
• File type:文件类型;
• Encoding:测序平台的版本和相应的编码版本号;
• Total Sequence:总 reads 数;
• Sequences flagged as poor quality:低质量序列数量;
• Sequence:测得的序列长度范围;
• %GC:GC 含量。
Per base sequence quality序列测序质量统计
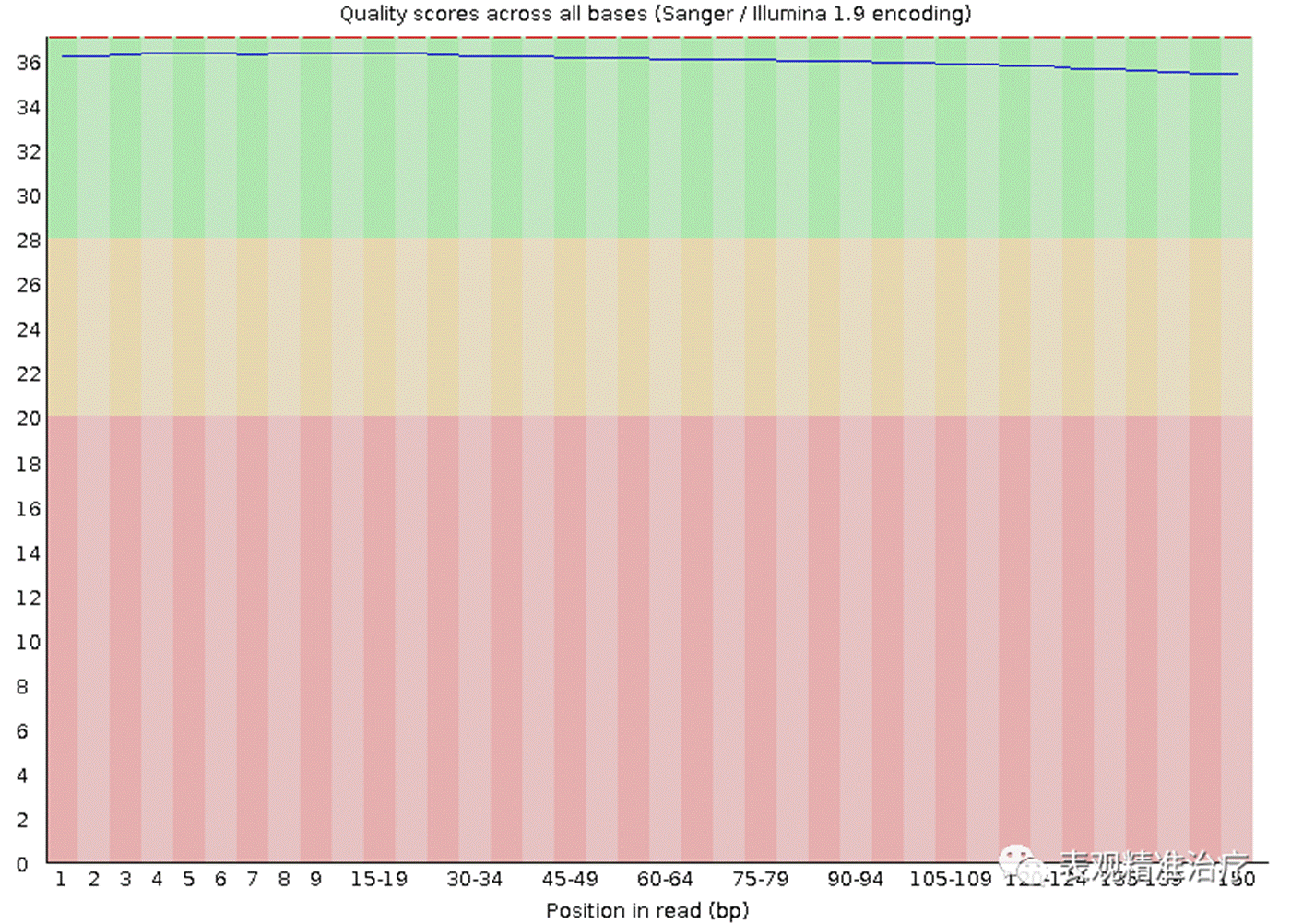
上图显示了检测 fastq 文件的整体碱基质量分数统计。
• 横轴表示测序文件中所有序列第一个碱基到最后一个碱基,纵轴表示质量得分;
• 红线表示中位数,蓝线代表平均值;
• 柱状表示该位置所有序列的测序质量的统计,柱状(黄色)是 25%~75% 区间质量分布,error bar(触须)是 10%~90% 区间质量分布;
• 一般要求所有位置的 10% 小于 20,即最多允许该位置 10% 的序列低于 Q20,即 90% 的序列的碱基质量都大于 Q20,即 90% 的序列碱基错误率不超过 99%。当任何碱基质量低于 10,或者任何中位数低于 25 时报 WARN;当任何碱基质量低于 5 或者任何中位数低于 20 报 FAIL。
Per tile sequence quality每个tail测序的情况
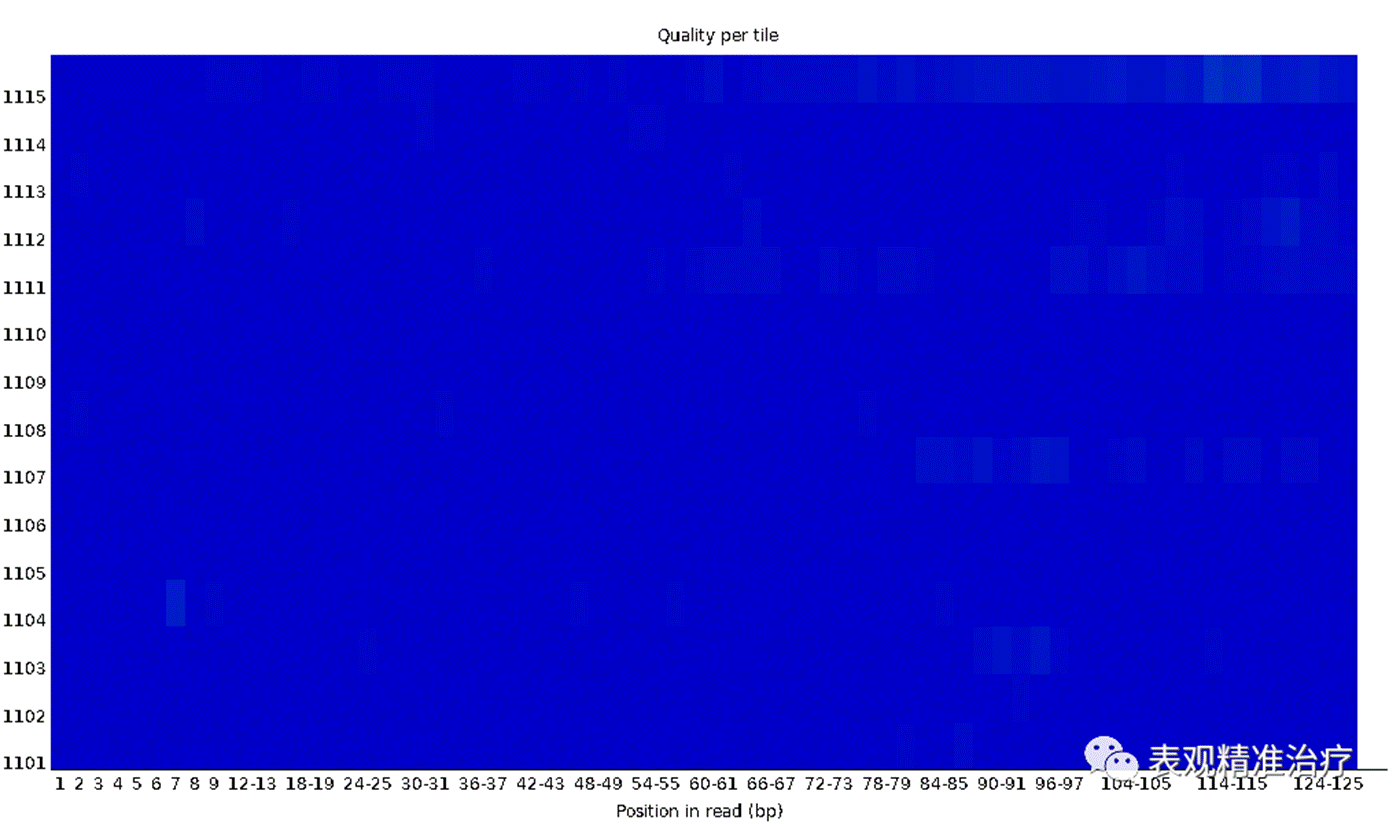
上图展示了每个 tail 的测序情况。
• 横轴表示每个碱基的位置;
• 纵轴是 tail 的 Index 编号;
• 这个图主要是为了防止,在测序过程中,某些 tail 受到不可控因素的影响而出现测序质量偏低;
• 蓝色代表测序质量很高,暖色代表测序质量不高,如果某些 tail 出现暖色,可以在后续分析中把该 tail 测序的结果全部都去除。
Per sequence quality scores每条序列的测序质量统计
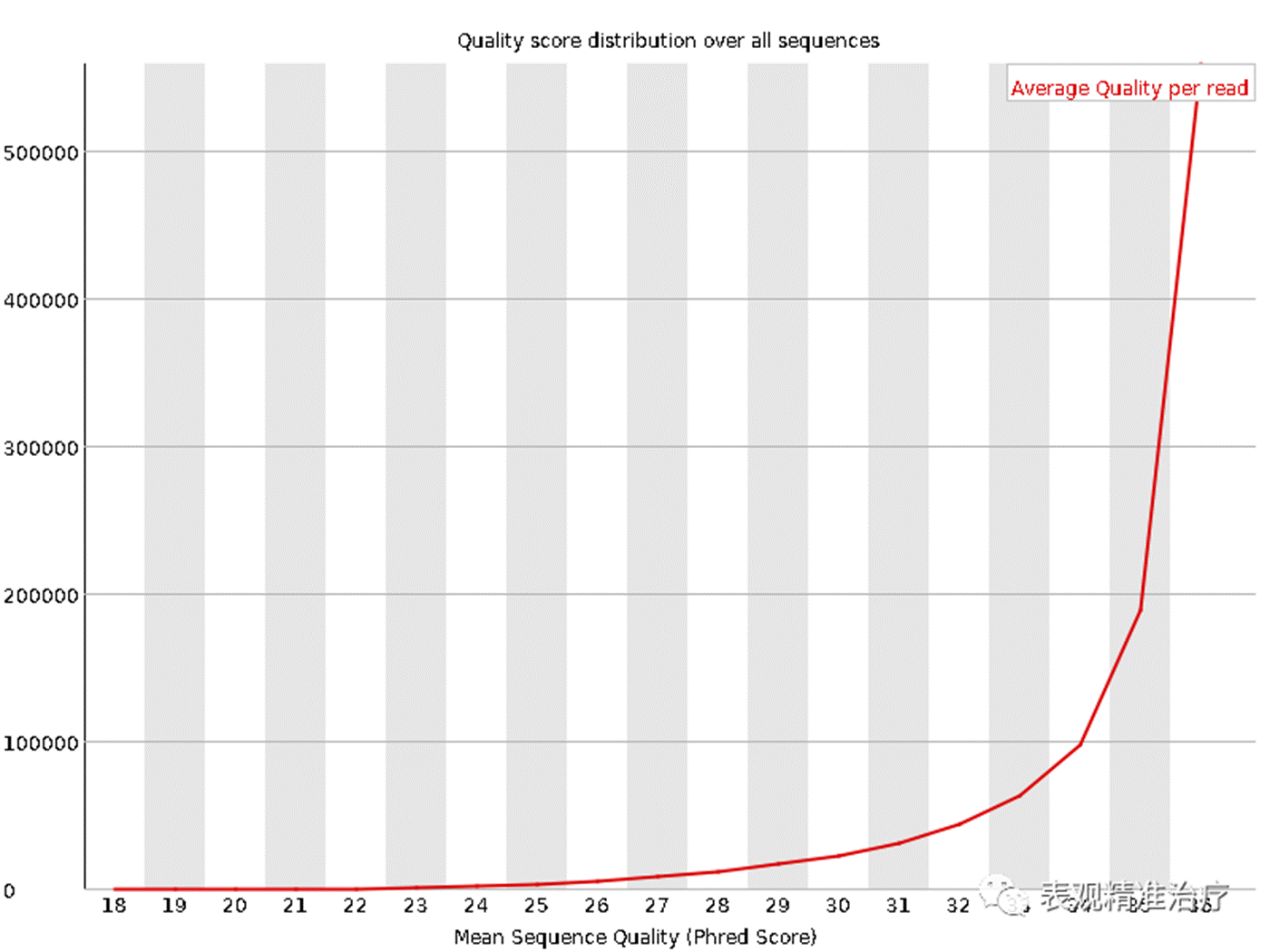
对每条序列(reads)的测序质量统计。
• 假如我测的 1 条序列长度为 101bp,那么这 101 个位置每个位置 Q 值的平均值就是这条 reads 的质量值;
• 该图横轴是 0-40,表示 Q 值,即该序列(reads)质量得分;
• 纵轴是每个值对应的 reads 数目;
• 我们的数据中,测序结果主要集中在高分中,证明测序质量良好。
Per base sequence content序列各个位置碱基比例分布
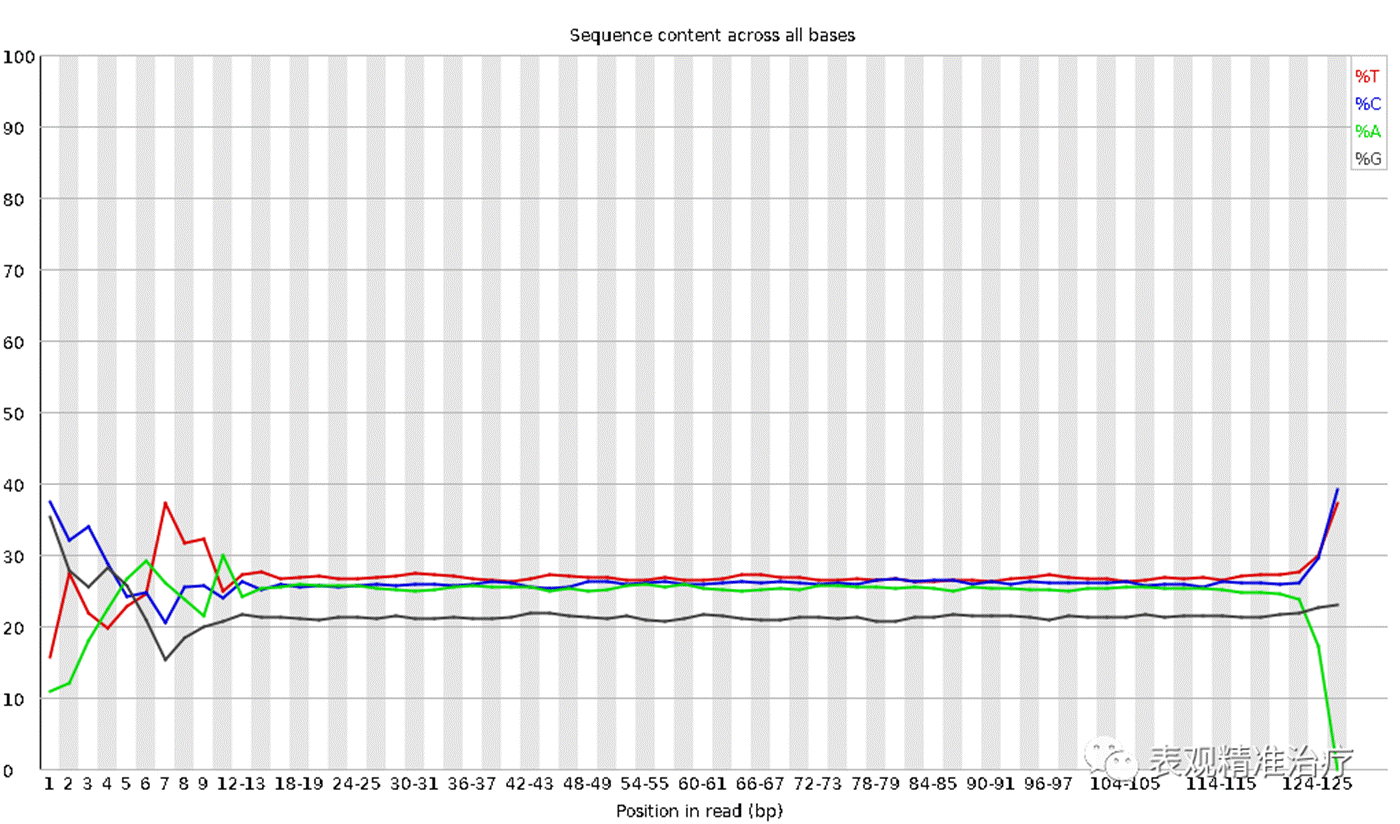
上图显示了 A T C G 在每个位置的平均分布情况。
• 横轴表示每个碱基的位置,纵轴表示百分比;
• 图中四条线代表 A T C G 在每个位置平均含量;
• 理论上来说,A 和 T 应该相等,G 和 C 应该相等,但是一般测序的时候,刚开始测序仪状态不稳定,很可能出现上图的情况。像这种情况,即使测序的得分很高,也需要 cut 开始部分的序列信息。
Per sequence GC content序列平均GC分布
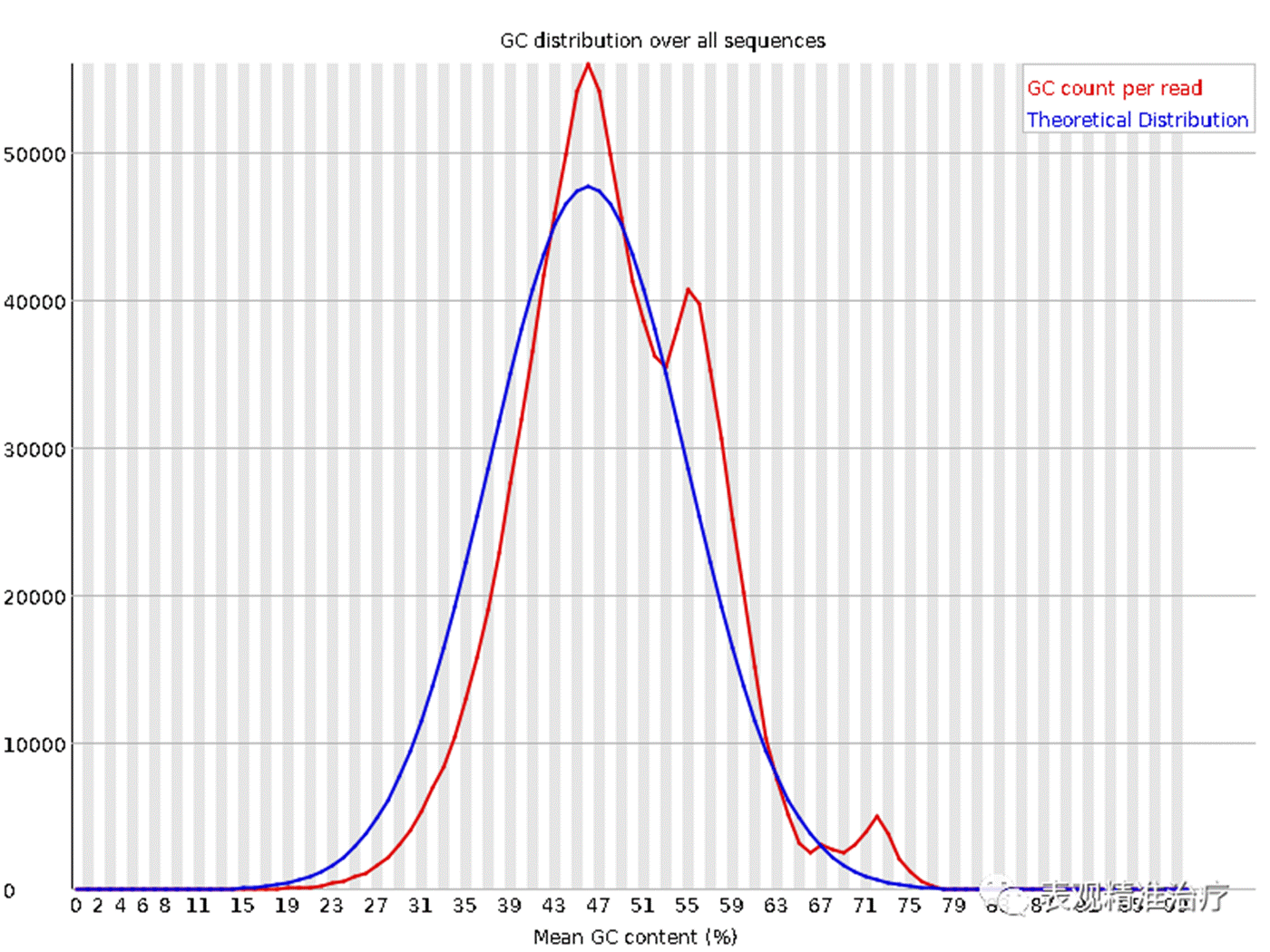
上图展示了序列平均 GC 分布。
• 横轴为平均 GC 含量;纵轴为每个 GC 含量对应的序列数量;
• 蓝线为系统计算得到的理论分布;红线为测量值,二者越接近越好;
• 这里不相符可能有两个原因:
1. GC 可以作为物种特异性根据,如果出现了其他的峰则提示有可能混入了其他物种的 DNA;
2. 目前二代测序基本都会有序列偏向性 (所说的 bias),也就是某些特定区域会被反复测序,以至于高于正常水平,变相说明测序过程不够随机。这种现象会对以后的变异检测以及 CNV 分析造成影响。
Per base N content N碱基含量分布
上图 N 碱基含量分布
• N 碱基是指仪器不能识别的碱基,一般不会出现。但是如果出现并且量还很大,应该就是测序系统或者试剂的问题;
• 任意位置的 N 的比例超过 5%,报 "WARN";任意位置的 N 的比例超过 20%,报 "FAIL"。
Sequence Length Distribution序列测序长度统计
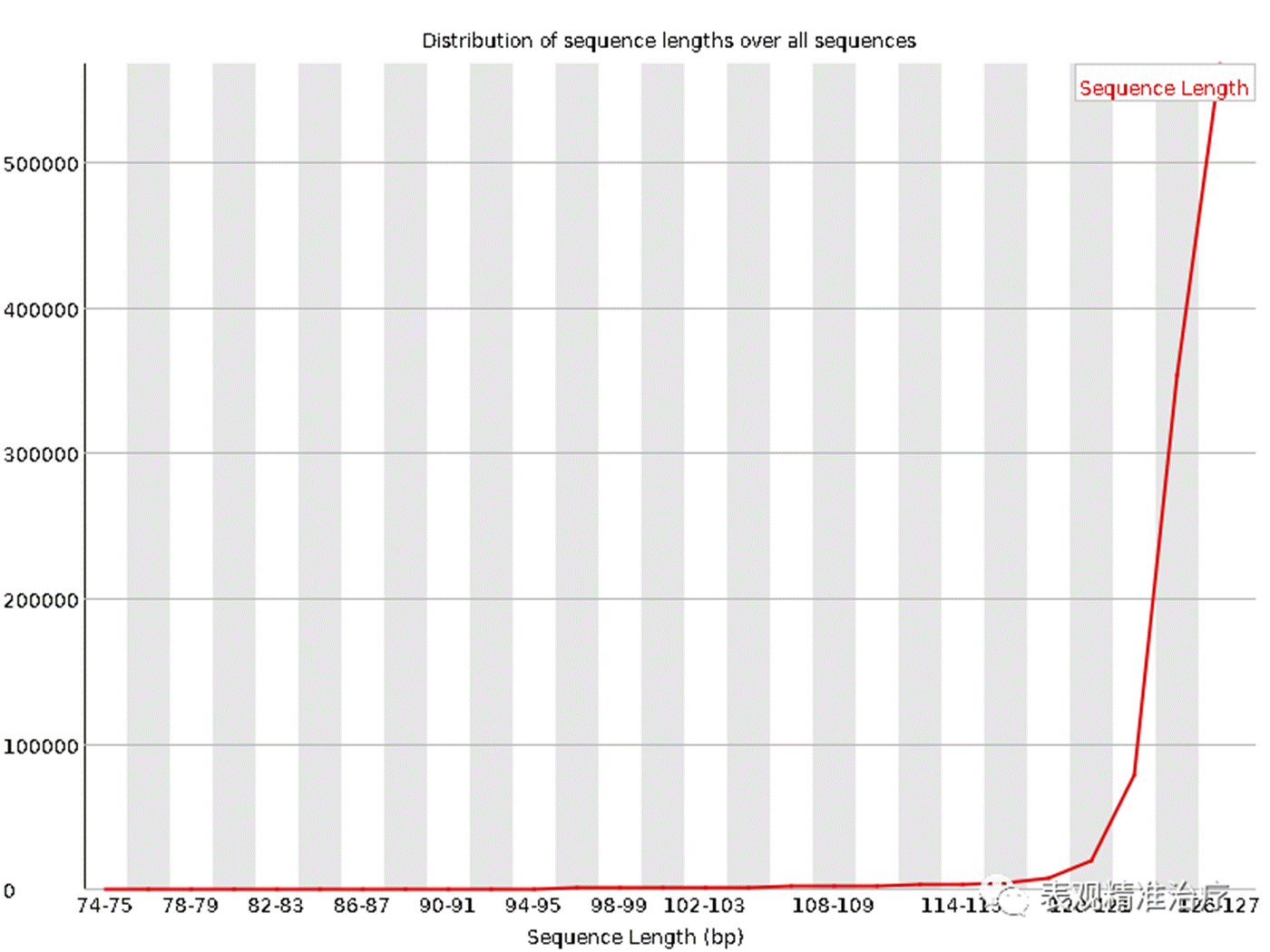
上图展示了检验文件中序列的长度统计。
• 每次测序仪测出来的长度在理论上应该是完全相等的,但是总会有一些偏差;
• 比如此图中,126-127bp 是主要的,但是还是有少量的 120-121bp 的长度,不过数量比较少,不影响后续分析;
• 当测序的长度不同时,如果很严重,则表明测序仪在此次测序过程中产生的数据不可信。
Sequence Duplication Levels统计序列完全一样的reads的频率
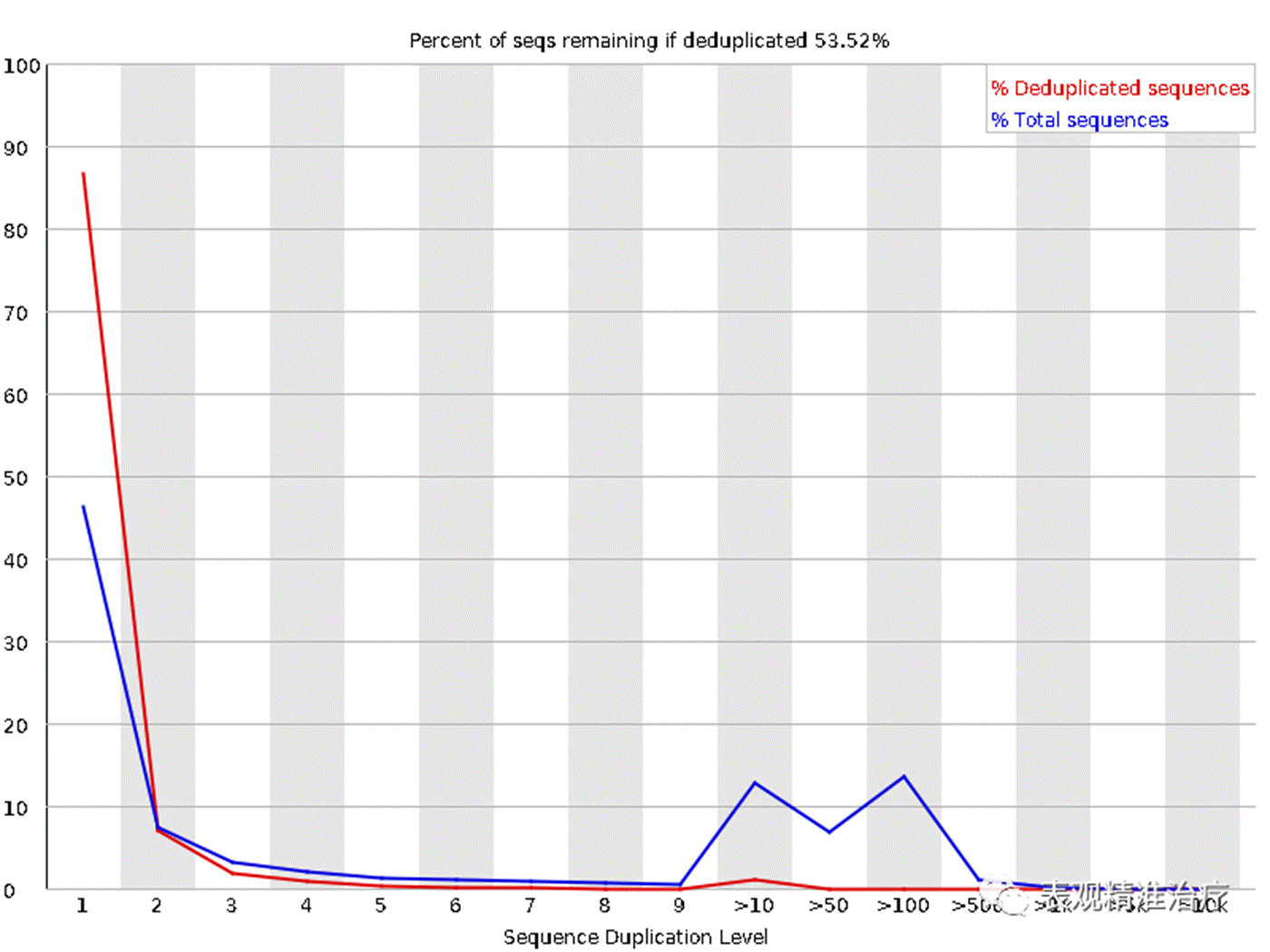
• 谈到 NGS 数据的 duplicated reads(暂且翻译为 “重复数据”),我们通常会直观地认为:duplicated reads 是在 NGS 文库构建过程中,由于 PCR 过度扩增导致同一个模板 DNA 片段被反复测序多次,得到一模一样的 reads;
• 上图中横坐标是 duplication 的次数;纵坐标是 duplicated reads 的数目(红线);
• 正常情况下的确,测序深度越高,越容易产生一定程度的 duplication。高程度的 duplication level,提示我们可能有 bias 的存在(如建库过程中的 PCR duplication)。
Overrepresented sequences大量重复序列
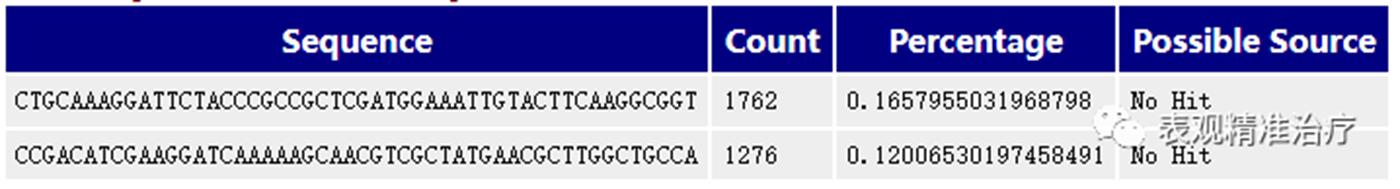
• Overrepresented sequences 是指一条序列的重复数,因为一个转录组中有非常多的转录本,一条序列再怎么多也不太会占整个转录组的一小部分(比如 1%),如果出现这种情况,不是这种转录本巨量表达,就是样品被污染。这个模块列出来大于全部转录组 1% 的 reads 序列,但是因为用的是前 200,000 条,所以其实参考意义不大,完全可以忽略。
• 和 duplication 计算一样,取前 200,000 进行统计,大于 75bp 只取 50bp;
• 发现超过总 reads 数 0.1% 的 reads 时报”WARN“,当发现超过总 reads 数 1% 的 reads 时报”FAIL“;
Adapter Content序列Adapter
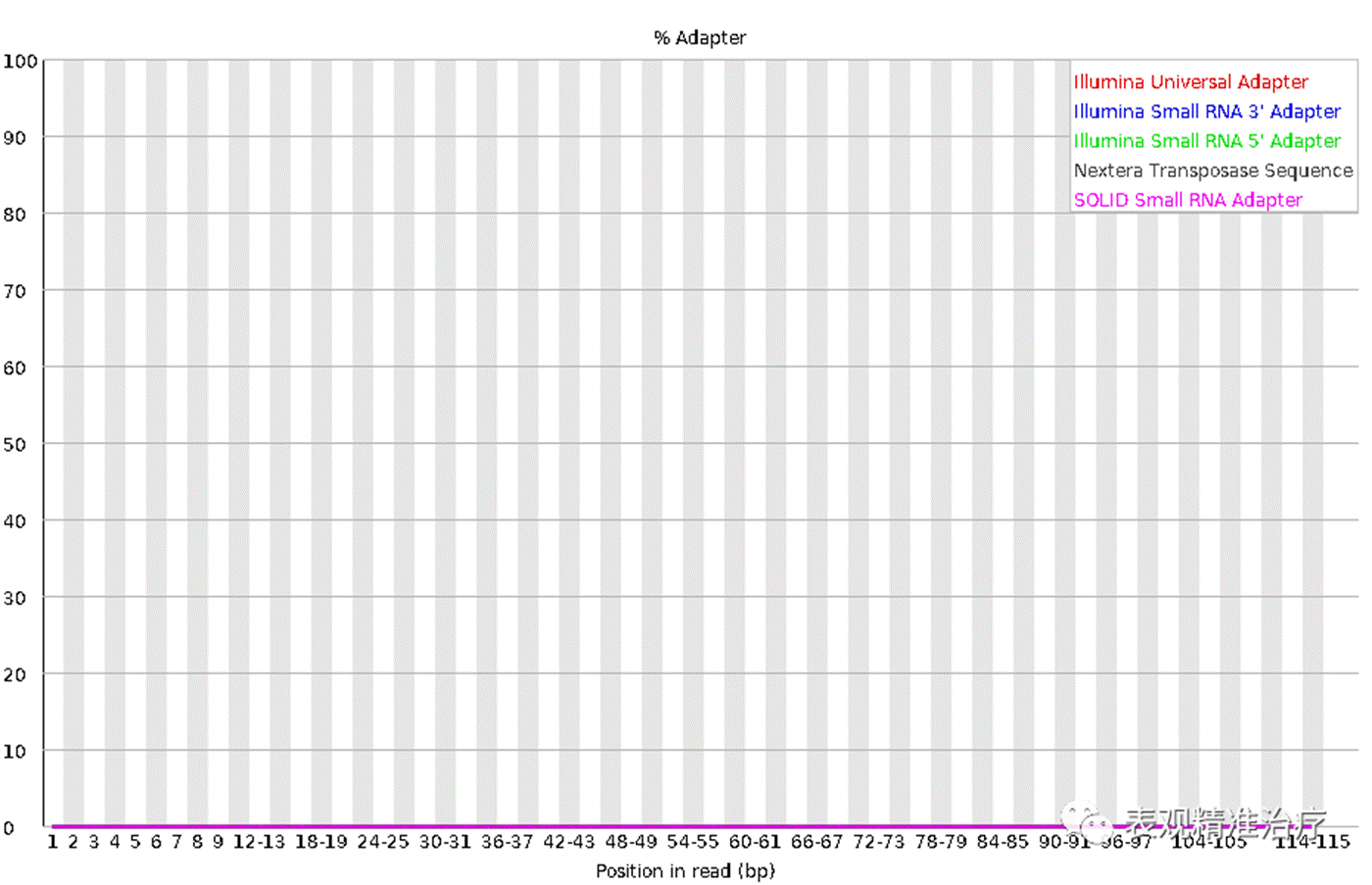
• 此图衡量的是序列中两端 adapter 的情况
• 如果在当时 fastqc 分析的时候 - a 选项没有内容,则默认使用图例中的四种通用 adapter 序列进行统计
• 本例中 adapter 都已经去除,如果有 adapter 序列没有去除干净的情况,在后续分析的时候需要先使用质控软件进行去接头。
接下来就是基于 QC 结果对数据进行质量控制,cutadapt、Trimmomatic、fastp 等许多软件都可以完成这一工作,随后会一一介绍。
关于测序数据的质量评估先介绍这么多,需要交流学习经验可以留言或添加下方小编微信,一起学习吧!!
小编微信号 : BioinforTec
图文:张皓旻
本文编辑:李晨龙

