本文由医信融合团队成员“张皓旻”撰写,已同步至微信公众号“医信融合创新沙龙”,更多精彩内容欢迎关注!

HTSeq作为一款可以处理高通量数据的python包,由Simon Anders, Paul Theodor Pyl, Wolfgang Huber等人携手推出HTSeq — A Python framework to work with high-throughput sequencing data。自发布以来就备受广大分析人员青睐,其提供了许多功能给那些熟悉python的大佬们去自信修改使用,同时也兼顾着给小白们提供了两个可以拿来可用的可执行文件htseq-count(计数)和htseq-qa(质量分析)。本文测试数据使用GSE176393。
HTseq使用要点:
这里需要注意的是HTSeq作为read counts的计数软件,承接的是上游比对软件对于clean data给出的比对结果,而且必须为依据reads名称排序的sort bam文件。
所以在之前需应用samtools对bam文件进行排序。
nohup cat ../SRR_Acc_List.txt | while read line; do samtools sort -o $line\_sorted.bam -n -T sorted $line.bam; done &(依然是批量后台运行)
(samtool是非常重要的生物信息软件,别急,走完转录组的常规分析流程我们就专门进行介绍)
下面开始今天的HTseq使用!!
安装
依然是使用conda进行安装,方便省事。conda install -c bioconda htseq=1.99.2
强调一下使用conda安装软件的时候一定要注明- c(使用的channel)及软件版本号,否则很多时候下载下来的软件并不能在WSL中直接使用。想要知道各个软件在conda中的最新版本号,在google中用“软件名conda”进行检索就可以很方便的获取。
定量
=========
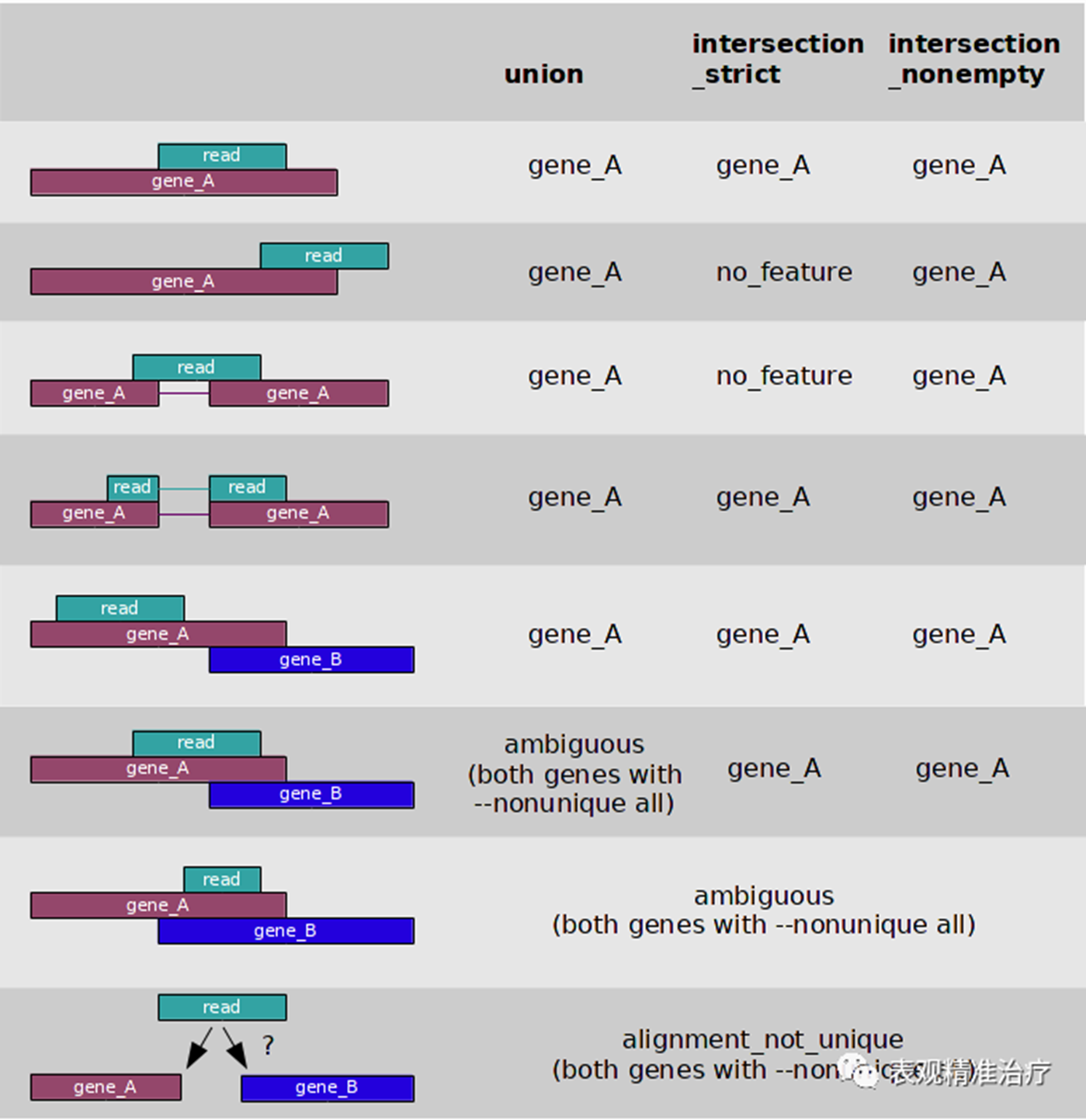
HTSeq计算counts数有3种模式,如下图所示(ambiguous表示该read比对到多个gene上;no_feature表示read没有比对到gene上):
此外,htseq-count对链特异性及非链特异性数据的处理是不同的因此在处理数据前一定要明确目标数据的建库方式(链特异性或非链特异性的),对于什么是链特异性和非链特异性以及具体的检验方式随后也会专门讲解(又是一个待填坑
 )。明确这一关键问题之后就可以进行定量啦!
)。明确这一关键问题之后就可以进行定量啦!
先看一下HTseq的常用参数:
#htseq-count常用参数
-f | --format default: sam
设置输入文件的格式,该参数的值可以是sam或bam。
-r | --order default: name
设置sam或bam文件的排序方式,该参数的值可以是name或pos。前者表示按read名进行排序,后者表示按比对的参考基因组位置进行排序。若测序数据是双末端测序,当输入sam/bam文件是按pos方式排序的时候,两端reads的比对结果在sam/bam文件中一般不是紧邻的两行,程序会将reads对的第一个比对结果放入内存,直到读取到另一端read的比对结果。因此,选择pos可能会导致程序使用较多的内存,它也适合于未排序的sam/bam文件。而pos排序则表示程序认为双末端测序的reads比对结果在紧邻的两行上,也适合于单端测序的比对结果。很多其它表达量分析软件要求输入的sam/bam文件是按pos排序的,但HTSeq推荐使用name排序,且一般比对软件的默认输出结果也是按name进行排序的。
-s | --stranded default: yes
设置是否是链特异性测序。该参数的值可以是yes,no或reverse。no表示非链特异性测序;若是单端测序,yes表示read比对到了基因的正义链上;若是双末端测序,yes表示read1比对到了基因正义链上,read2比对到基因负义链上;reverse表示双末端测序情况下与yes值相反的结果。根据说明文件的理解,一般情况下双末端链特异性测序,该参数的值应该选择reverse(本人暂时没有测试该参数)。
-a | --a default: 10
忽略比对质量低于此值的比对结果。在0.5.4版本以前该参数默认值是0。
-t | --type default: exon
程序会对该指定的feature(gtf/gff文件第三列)进行表达量计算,而gtf/gff文件中其它的feature都会被忽略。
-i | --idattr default: gene_id
设置feature ID是由gtf/gff文件第9列那个标签决定的;若gtf/gff文件多行具有相同的feature ID,则它们来自同一个feature,程序会计算这些features的表达量之和赋给相应的feature ID。
-m | --mode default: union
设置表达量计算模式。该参数的值可以有union, intersection-strict and intersection-nonempty。这三种模式的选择请见上面对这3种模式的示意图。从图中可知,对于原核生物,推荐使用intersection-strict模式;对于真核生物,推荐使用union模式。
-o | --samout
输出一个sam文件,该sam文件的比对结果中多了一个XF标签,表示该read比对到了某个feature上。
-q | --quiet
不输出程序运行的状态信息和警告信息。
-h | --help
输出帮助信息。
通过对数据的检测,我的数据是一个非链特异性的数据,所以进行下面操作:
#我的批量分析代码
#写个bash
vim htseq.sh
cat SRR_Acc_List.txt | while read line
do
htseq-count -f bam -s no -m intersection-nonempty 2-mapping/$line\_sorted.bam ../../reference/homo/Homo_sapiens.GRCh38.104.gtf > 3-quantity/$line\_count.txt
done
#后台运行bash
nohup bash htseq.sh &
获得如下结果:

发现一个问题,每个样本的定量结果都在单独的文件中,但是一般我们做后续分析需要把各个样本的表达量放在一个文件中,所以我们利用R语言对数据整理一下。
数据整理
我们利用R语言对结果进行合并,先看一下文件内容
##看一下任意文件前十行
head SRR14760842_count.txt
ENSG00000000003 6157
ENSG00000000005 0
ENSG00000000419 2925
ENSG00000000457 791
ENSG00000000460 910
ENSG00000000938 0
ENSG00000000971 2192
ENSG00000001036 3245
ENSG00000001084 1427
ENSG00000001167 1897
#看一下任意文件后十行
tail SRR14760842_count.txt
ENSG00000288721 6
ENSG00000288722 166
ENSG00000288723 1
ENSG00000288724 0
ENSG00000288725 1
__no_feature 1980267
__ambiguous 1633742
__too_low_aQual 821454
__not_aligned 961622
__alignment_not_unique 1571191
观察数据我们可以发现,最后五行是不需要的因此需要去除,此外文件缺少表头信息,所以在处理时要注意标注,同时我们还需要对ensembleid进行注释。
明确了目的就开始处理吧!
setwd("F:\\RNA-Seq\\GSE176393(SRP323246)\\3-quantity") #HTseq结果文件目录,可修改
library(magrittr)
library(clusterProfiler)
library(org.Hs.eg.db)
library(limma)
#读取文件
files = list.files(pattern = "_count")
file <- list()
for (i in files) {
file[[i]]<-read.table(i,sep = "\t",check.names = F,col.names = c("gene_id",i))
}
rt = Reduce(function(x,y) merge(x,y,by="gene_id",all.x=TRUE),file,accumulate=FALSE) # R语言魔法,一句话合并
#注释
rt <- rt[-1:-5,]
ensembl = rt[,1]
geneSymbol <- bitr(ensembl, fromType = "ENSEMBL", #fromType为输入数据ID类型
toType = "SYMBOL", #toType是指目标类型
OrgDb = org.Hs.eg.db)#Orgdb是指对应的注释包,这是人的注释包,相关注释包请更换
rt <- merge(geneSymbol,rt,by.x="ENSEMBL",by.y="gene_id")
#对基因名称重复的reads count进行处理
rt<-rt[,-1]
rt<-as.matrix(rt)
rownames(rt)<-rt[,1]
rt<-rt[,2:ncol(rt)]
rt<-avereps(rt)
rt1<-apply(rt,2,as.numeric)
rownames(rt1)<-rownames(rt)
rt1<-as.data.frame(rt1)
rt1<-cbind(row.names(rt1),rt1)
colnames(rt1)[1]<-"genes"
#输出
write.table(rt1,"rawreadcount.txt",sep = "\t",row.names = F,col.names = T,quote = F)
最后就获得了合并的read count文件
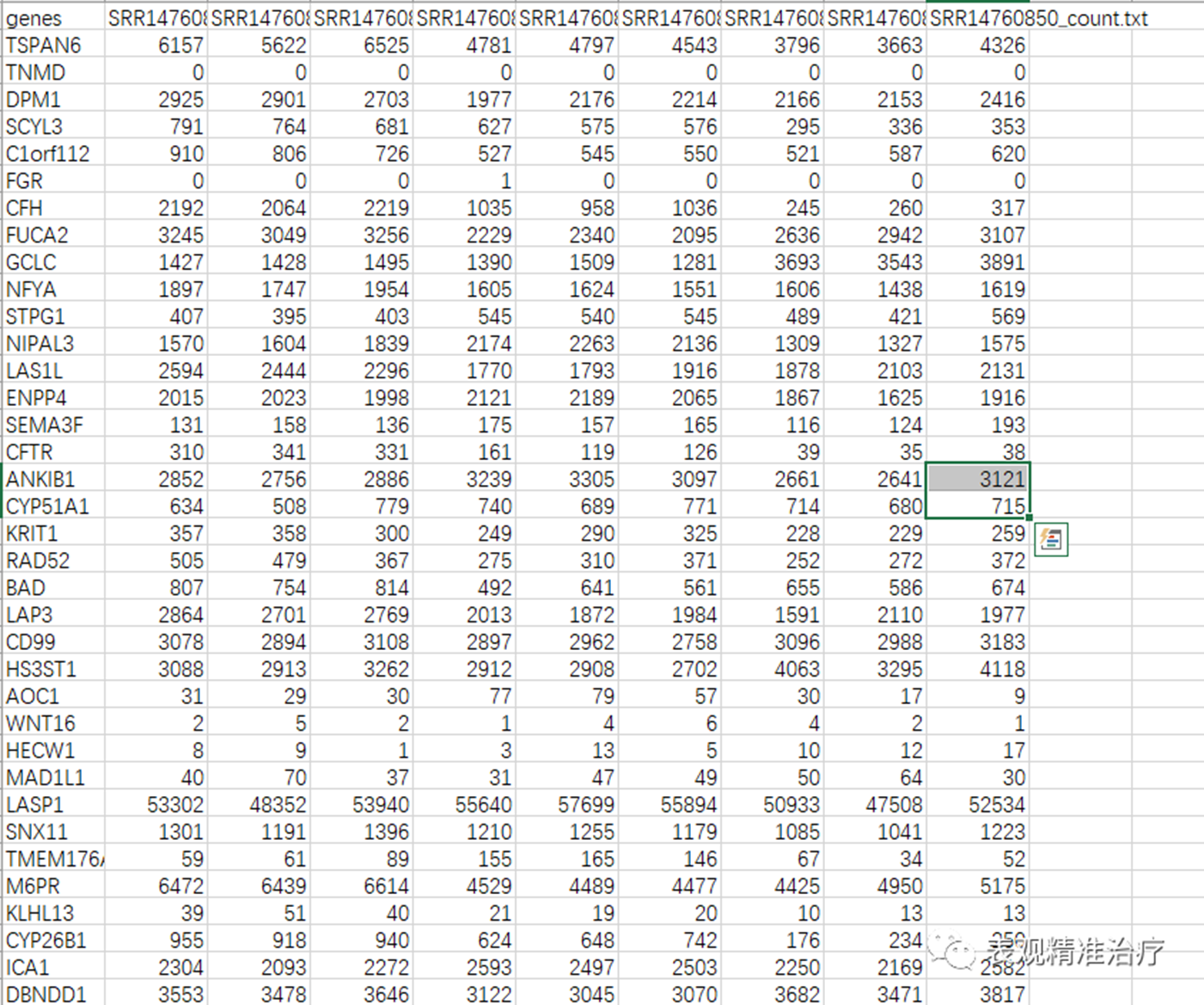
整理成这样,做过芯片数据的小伙伴是不是就很眼熟啦!
是不是很简单,快去自己的电脑上试一试吧!!
图文:张皓旻
本文编辑:李新龙

