本文由医信融合团队成员“张皓旻”撰写,已同步至微信公众号“医信融合创新沙龙”,更多精彩内容欢迎关注!

Salmon是转录本定量软件,使用转录本定量主要优点一是更准确,比如不同样本同一基因使用不同isoform此时基因长度并不相等,直接基因定量无法顾及;二是方便进行可变剪切分析。Salmon提供2种运行模式,一是直接读取reads文件(姑且称为fastq模式);二是读取比对好文件sam/bam (姑且称为比对模式)。
salmon是一款不基于比对而直接对基因进行定量的工具,适用于转录组、宏基因组等分析。
其优势是:
• 定量时考虑到不同样品中基因长度的改变(比如不同isoform的使用)
• 速度快、需要的计算资源和存储资源小
• 敏感性高,不会丢弃匹配到多个基因同源区域的reads
• 可以直接校正GC-bias
• 自动判断文库类型
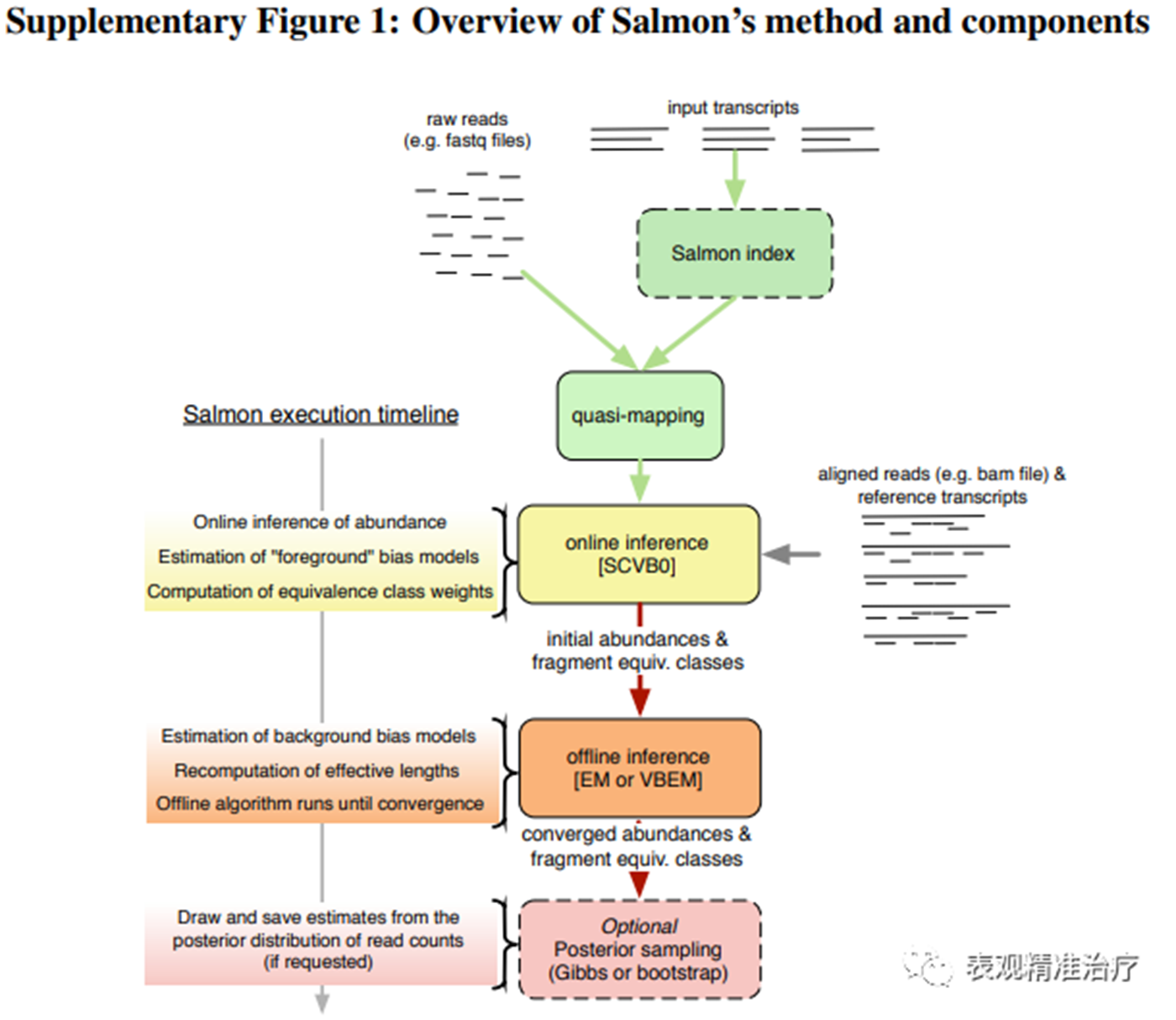
salmon的定量依赖于cDNA序列(参考转录组)、基因组注释文件以及原始fastq文件。
使用conda进行安装conda install -c bioconda salmon=1.6.0,安装完后看一下help文档。
salmon -h
salmon v1.6.0
Usage: salmon -h|--help or
salmon -v|--version or
salmon -c|--cite or
salmon [--no-version-check] <COMMAND> [-h | options]
Commands:
index : create a salmon index
quant : quantify a sample
alevin : single cell analysis
swim : perform super-secret operation
quantmerge : merge multiple quantifications into a single file
1构建索引
进行salmon定量前需要构建salmon index,构建index所需要的文件是参考转录组,运行以下命令:
salmon index -t /mnt/f/reference/homo/Homo_sapiens.GRCh38.cdna.all.fa -i homo
构建好的index长这样
2定量
接下来,就可以对数据进行定量啦!
定量前,有一个重要的参数,需要明确,就是- l参数的设置,即你的数据的建库方式,官方给出了这样的介绍:
Salmon, has the user provide a description of the type of sequencing library from which the reads come, and this contains information about e.g. the relative orientation of paired-end reads. As of version 0.7.0, Salmon also has the ability to automatically infer (i.e. guess) the library type based on how the first few thousand reads map to the transcriptome. To allow Salmon to automatically infer the library type, simply provide -l A or --libType A to Salmon. Even if you allow Salmon to infer the library type for you, you should still read the section below, so that you can interpret how Salmon reports the library type it discovers.
The first part of the library string (relative orientation) is only provided if the library is paired-end. The possible options are:
I = inward
O = outward
M = matching
The second part of the read library string specifies whether the protocol is stranded or unstranded; the options are:
S = stranded
U = unstranded
If the protocol is unstranded, then we’re done. The final part of the library string specifies the strand from which the read originates in a strand-specific protocol — it is only provided if the library is stranded (i.e. if the library format string is of the form S). The possible values are:
F = read 1 (or single-end read) comes from the forward strand
R = read 1 (or single-end read) comes from the reverse strand
An example of some library format strings and their interpretations are:
IU (an unstranded paired-end library where the reads face each other)
SF (a stranded single-end protocol where the reads come from the forward strand)
OSR (a stranded paired-end protocol where the reads face away from each other,
read1 comes from reverse strand and read2 comes from the forward strand)
在不清楚建库类型的情况下可以让salmon自动推断,-l A或者--libType A但是这种方式总感觉不踏实,所以我选择提前弄清楚,这就用到了,这个文章“如何鉴定数据的建库方式”(快去学习吧)。
通过鉴定,我的数据是一个非链特异性的双端测序数据,因此我需要使用如下命令:
salmon quant -i <index path> -l IU -1 <reads1 file> -2 <reads2 file> --validateMappings --gcbias --seqbias -o out
#批量运行脚本
cat SRR_Acc_List.txt | while read line
do
salmon quant -i /mnt/f/index/salmon_index/homo -l IU -1 ./1-cleandata/$line\_clean_1.fastq.gz -2 ./1-cleandata/$line\_clean_2.fastq.gz --validateMappings --gcBias --seqBias -g /mnt/f/reference/homo/Homo_sapiens.GRCh38.104.gtf -o ./3-salmon/$line
done
##命令解释
-i index目录位置
-l文库类型
-1 read1文件名称
-2 read2文件名称
--validateMappings对mapping到参考转录组的reads进行矫正,提高准确性,但会增加运算量
--gcBias对GC含量进行矫正,提高准确性,但会增加运算量
--seqBias矫正“sequence-specific bias”,提高准确性,但会增加运算量
-g #提供转录本与基因之间对应关系的文件。如果提供了该文件,Salmon将会输出文件:quant.sf和quant.genes.sf。
#其中quant.genes.sf文件中含有对基因表达水平的估计。
#提供的文件可以是GTF文件,也可以是由TAB分隔符分隔文件,文件中的每一行含转录本名称和对应的基因名。
#以'.gtf', '.gff'或'.gff3'结尾的文件名均被当做GTF格式做处理解析。
#以其他字符结尾的被当做由TAB分隔符分隔的简单文件格式做处理解析。
-o输出文件目录名称
接下来你会得到如下结果。
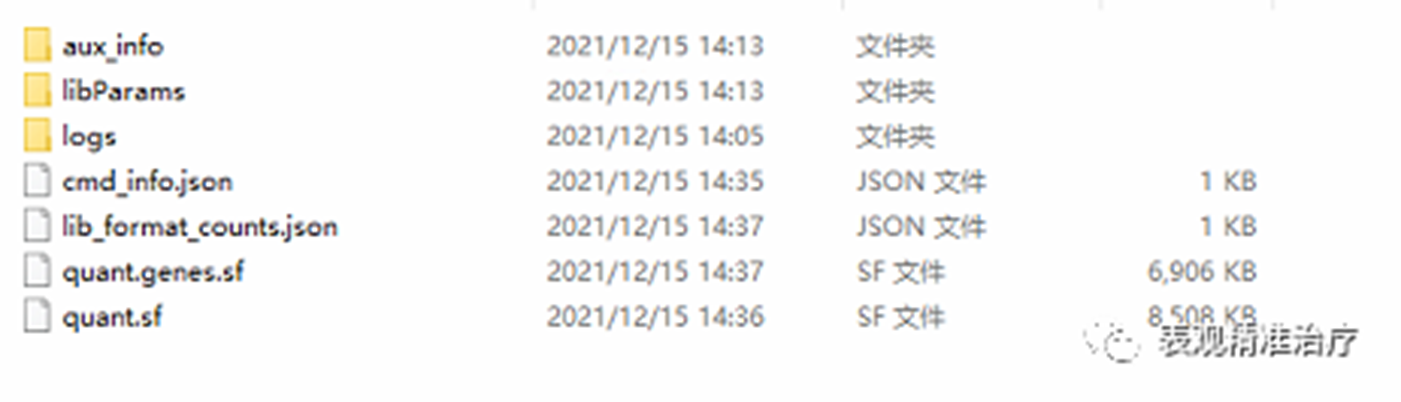
结果文件解读:
aux_info:其中包含很多文件,元数据信息(meta_info.json)、Unique and ambiguous count file(ambig_info.tsv)、Fragment length distribution(fil.gz)等
libParams:表达值tsv结果文件。包括转录本ID,length,eff_length, est_counts,tpm等。
logs:日志文件
cmd_info.json:运行使用的命令行参数
lib_format_counts.jsom:这个JSON文件报告至少有一个映射与指定的库格式兼容的片段数量,以及不兼容的片段数量
quant.genes.sf:基因表达定量结果(reads count和TPM)
quant.sf:转录本表达定量结果(reads count和TPM)
salmon运行速度很快,也很方便,对于不进行结构分析想最快获取表达谱数据的生信人来说最适合不过,快去试试吧。
后记
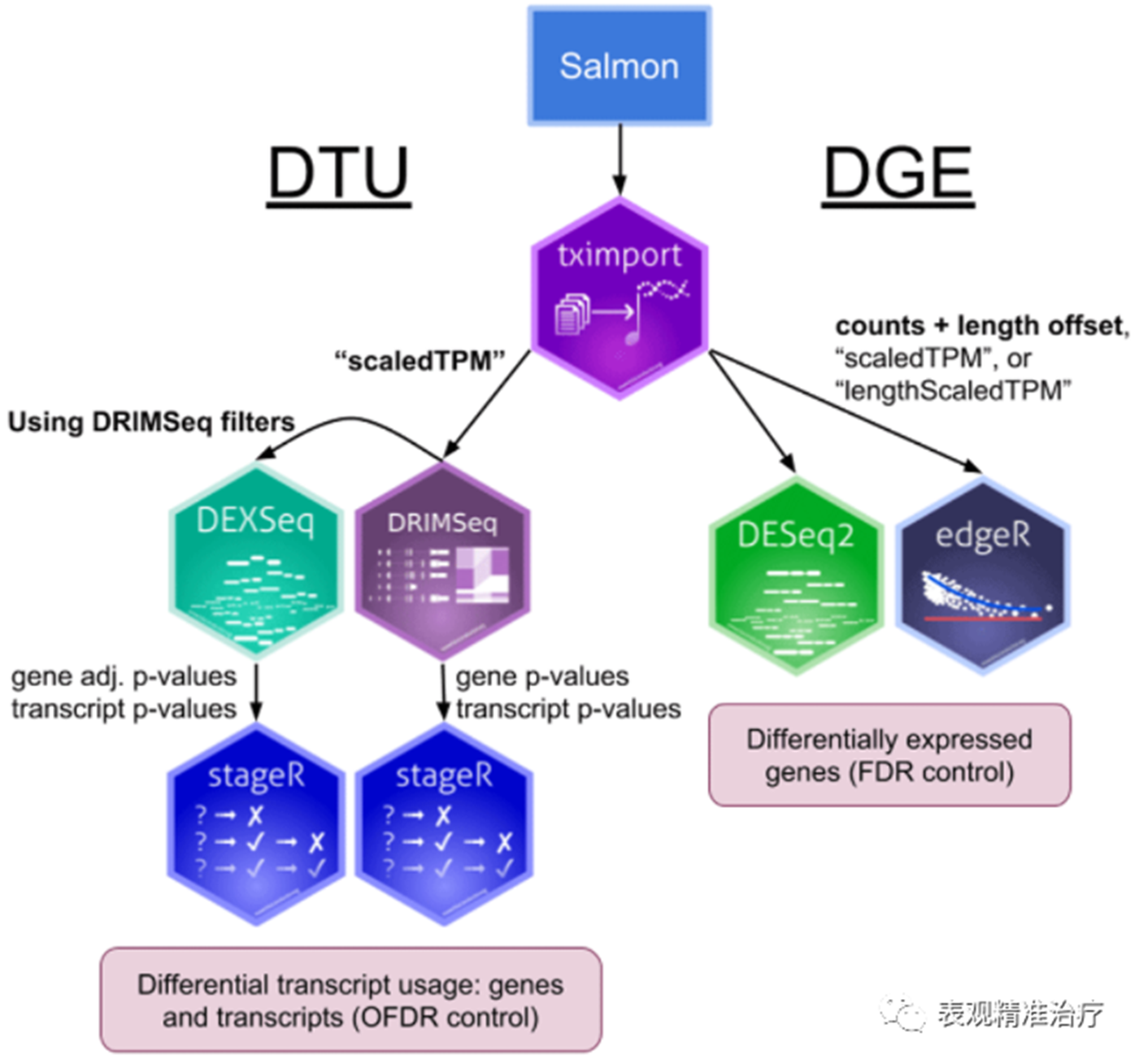
salmon定量后就是差异分析了,但是思路与常规的使用reads count进行差异分析有些不同。因为他是对转录本的定量,所以其准确性相较于基因水平的定量要准确许多,后续差异分析上自然也分为转录本水平的差异分析和基因水平的差异分析,两者结合则更可以全面评估基因在表达方面的变化,具体思路如下图:
图文:张皓旻
本文编辑:李新龙

