本文由医信融合团队成员“张皓旻”撰写,已同步至微信公众号“医信融合创新沙龙”,更多精彩内容欢迎关注!

今天尝试一下用DESeq和TMM方法对readcount进行标准化
(文末附完整代码)
RNA-seq定量得到每个基因或转录本原始的readcount值,除了基因真实表达水平,readcount值还受到其它因素的影响,主要是基因长度和测序深度。最简单的标准化方式是计算counts per million (CPM),即原始reads count除以总reads数乘以1,000,000。这种计算方式的缺点是容易受到极高表达且在不同样品中存在差异表达的基因的影响;这些基因的打开或关闭会影响到细胞中总的分子数目,可能导致这些基因标准化之后就不存在表达差异了,而原本没有差异的基因标准化之后却有差异了。RPKM、FPKM和TPM是CPM按照基因或转录本长度归一化后的表达,也会受到这一影响。
DESeq
DESeq主要使用量化因子的方法对readcount进行标准化,其方法是首先计算每个基因在所有样品中表达的几何平均值。每个样品的量化因子(size factor)是所有基因与其在所有样品中的表达值的几何平均值的比值的中位数。具体实现需要应用R包DESeq2进行,代码如下:
library(DESeq2) #导入包
setwd("***") #设置工作目录
a<-read.table("*.xls",header = T,sep = "\t") ##读入readcount数据
row.names(a)<-a[,1]
a<-a[,-1]
sampleTable <- data.frame(condition = factor(rep(c("control","Experimental"), each = 10))) ##设置实验设计
dds <- DESeqDataSetFromMatrix(countData = round(a), colData = sampleTable, design = ~condition)
dds <- DESeq(dds)
normalized_counts <- counts(dds, normalized=TRUE) ##获取标准化矩阵
write.table(normalized_counts,file= "normalized_DESeq.xls",sep = '\t',row.names = T,quote = F)
输入文件格式:
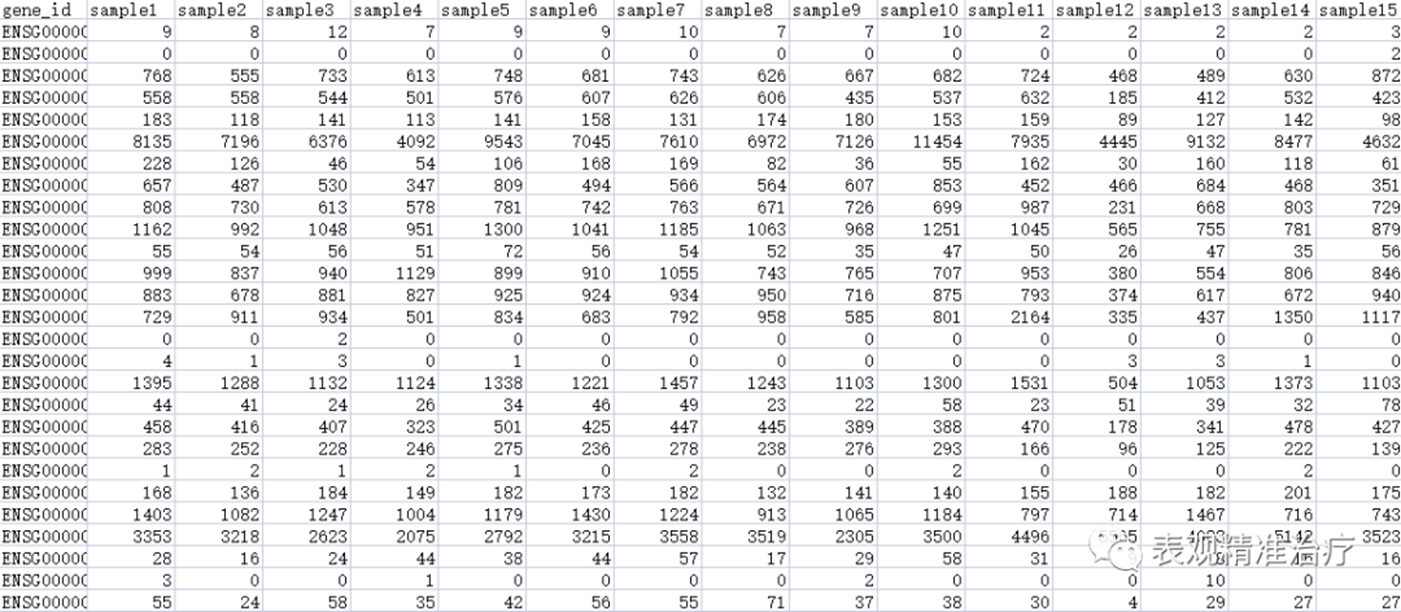
输出文件格式:
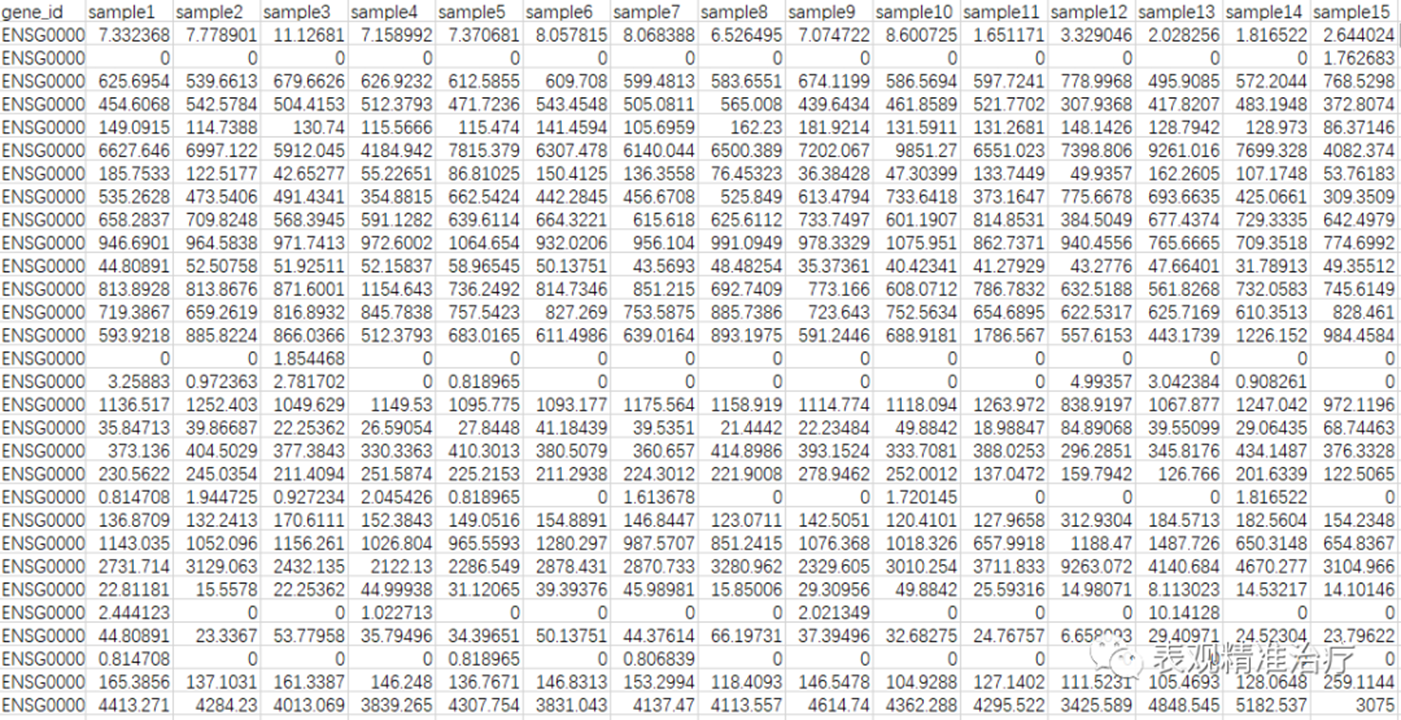
TMM
TMM是M -值的加权截尾均值。选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M -值。随后去除M -值中最高和最低的30%,剩下的M值计算加权平均值。每一个非参照样品的基因表达值都乘以计算出的TMM。这个方法假设大部分基因的表达是没有差异的。
具体实现需要应用R包edgeR实现,代码如下:
library(edgeR) ##导入包
setwd("***") ##设置工作目录
a<-read.table("*.xls",header = T,sep = "\t") ##读入readcount数据
row.names(a)<-a[,1]
a<-a[,-1]
group=c(rep("normal",10),rep("tumor",10)) ##设置实验设计
design <- model.matrix(~group)
dgelist <- DGEList(counts=a,group=group)
y<-calcNormFactors(dgelist,method = 'TMM')
c<-t(t(y$counts) / y$samples$lib.size / y$samples$norm.factors) * 1000000 ##获取标准化矩阵
write.table(c,file= "normalized_TMM.xls",sep = '\t',row.names = T,quote = F)
输入文件格式:
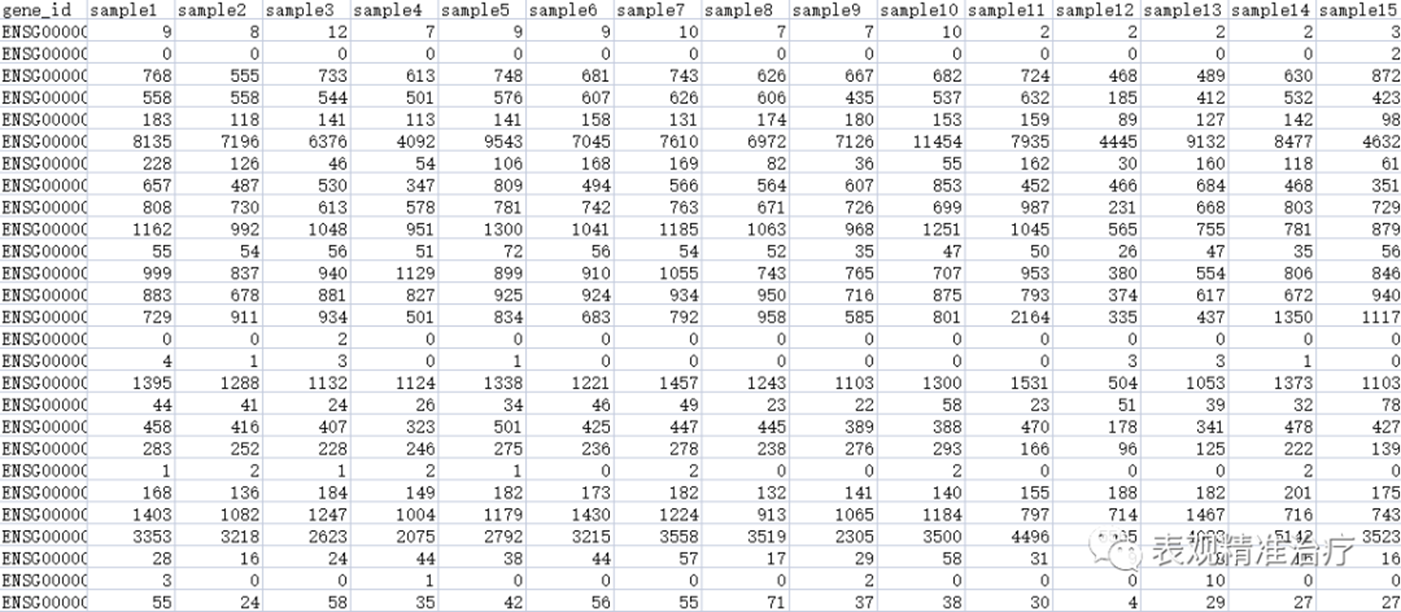
输出结果格式:

图文:张皓旻
本文编辑:李新龙

