在经过基因组组装或转录组差异基因表达量分析之后,对其结果进行注释是比较重要的一步,如何注释以及如何得到精确的注释结果?
Nr数据库,全称为Non-RedundantProtein Sequence Database,即包含了GenPept、Swissprot、PIR、PDF、PDB以及RefSeq Database中序列的蛋白质数据库。
(1)数据库下载
个人将下载方式分为图形界面下载和命令行方式下载。
需要使用的软件安装:
# blast
wget https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.12.0+-x64-linux.tar.gz
tar -zxvf ncbi-blast-2.12.0+-x64-linux.tar.gz
echo 'PATH=your_path/blast/ncbi-blast-2.12.0+/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
# aspera
wget -c https://d3gcli72yxqn2z.cloudfront.net/connect_latest/v4/bin/ibm-aspera-connect_4.1.0.46-linux_x86_64.tar.gz
tar -zxvf ibm-aspera-connect_4.1.0.46-linux_x86_64.tar.gz
#自动安装
sh ibm-aspera-connect_4.1.0.46-linux_x86_64.tar.gz
echo 'PATH=~/.aspera/connect/bin/:$PATH' >> ~/.bashrc
source ~/.bashrc
1、手动下载,即**图形界面下下载**
【标注】此处下载的数据为fasta格式,并非为pre-formatted(即提前构建好的Nr数据库)
步骤为:
(1)登录NCBI官网
(2)点击Download
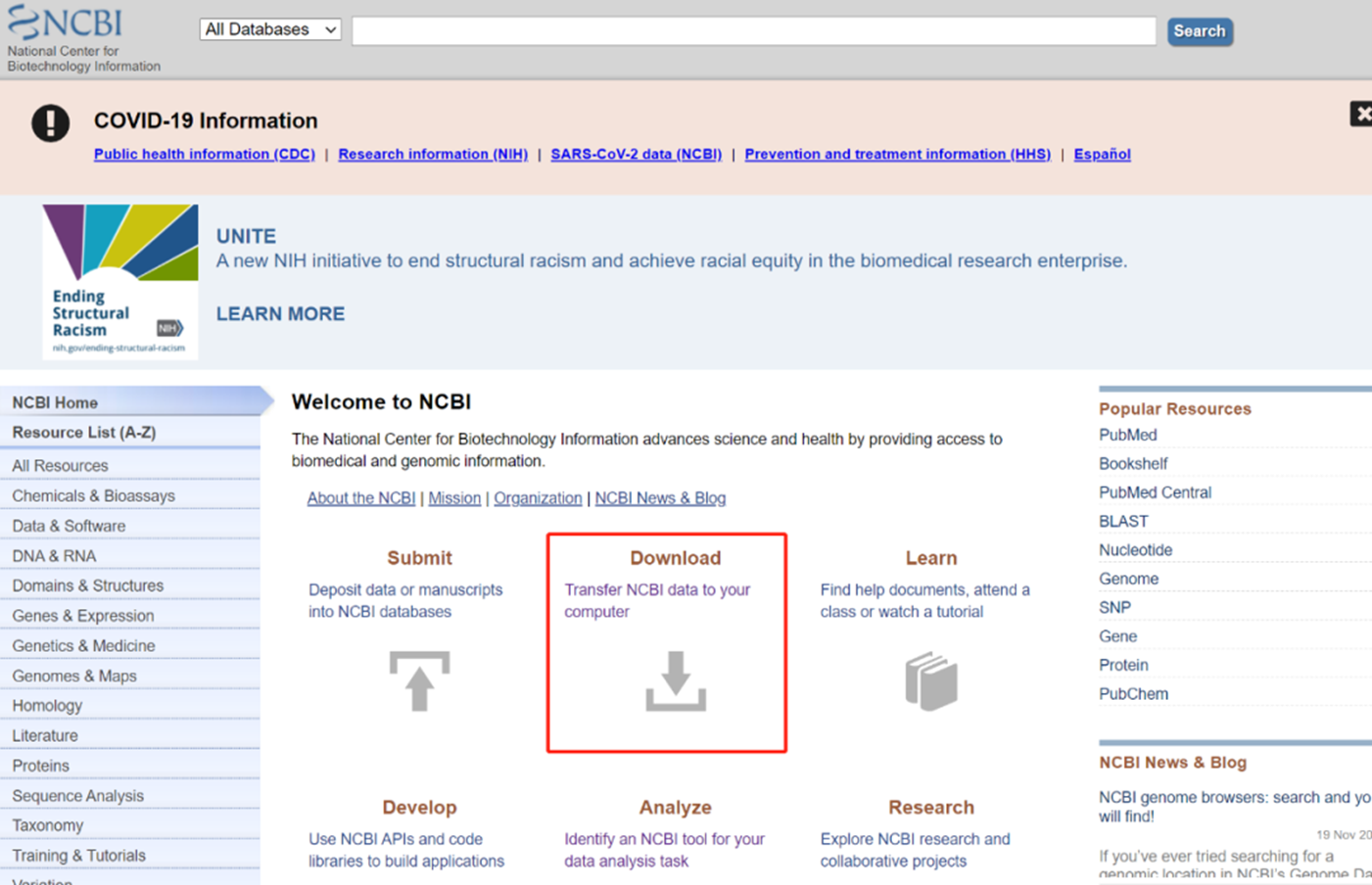
(3)点击FTP

(4)进入到对应路径下:https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/
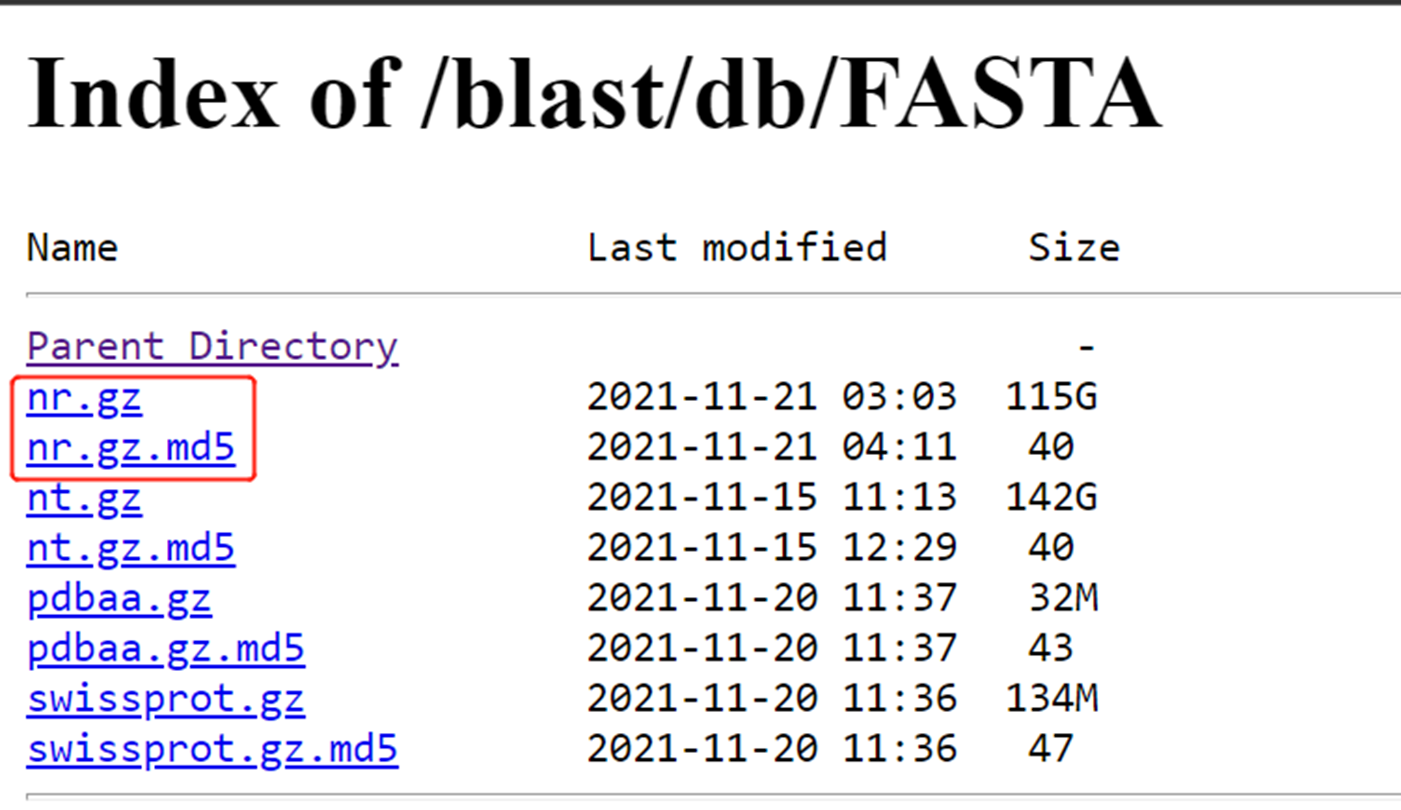
(5)右键复制nr.gz和nr.gz.md5的链接
(6)选择迅雷等工具下载即可
2、命令行界面下载
1. aspera
1.1下载nr.gz
ascp -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -l 200M -k 1 --overwrite=diff -QT anonftp@ftp.ncbi.nlm.nih.gov:blast/db/FASTA/nr.gz ./
#使用参数说明:
-i # Use public key authentication and specify the private key file.
#该密匙保存位置一般为:~/.aspera/connect/etc/
-l # Set the target transfer rate in Kbps. Default: 10000
#即设置最大传输速率
-k #可选参数为{0|1|2|3},即断点续传,一般设置为1
# Enable resuming partially transferred files. (Default: 0). Please note that this must be specified for your first transfer; otherwise, it will not work for subsequent transfers.
# “-k 1的具体说明”:-k 1 If the transfer stops because of connection loss or other issues the k option tells the transfer to resume from where it left off rather than restarting the entire transfer over.
###其他参数说明:
# 0 Always retransfer the entire file.
# 1 Check file attributes and resume if the current and original attributes match.
# 2 Check file attributes and do a sparse file checksum; resume if the current and original attributes/checksums match.
# 3 Check file attributes and do a full file checksum; resume if the current and original attributes/checksums match.
--overwrite=diff #默认设置即为diff,表示如果目前下载的文件与之前下载的文件不一致,则会overwrite
-Q # Enable fair (-Q) or trickle (-QQ) transfer policy. Use the -l and -m to set the target and minimum rates.
-T # Disable encryption for maximum throughput.
1.2下载preformatted版本
for i in {00..55}
do
echo "ascp -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -l 200M -k 1 --overwrite=diff -QT anonftp@ftp.ncbi.nlm.nih.gov:/blast/db/nr.${i}.tar.gz ./" >> aspera.preformatted.nr.sh
echo "ascp -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -l 200M -k 1 --overwrite=diff -QT anonftp@ftp.ncbi.nlm.nih.gov:/blast/db/nr.${i}.tar.gz.md5 ./" >> aspera.preformatted.nr.sh
done
ParaFly -c aspera.preformatted.nr.sh -CPU 1 &
#检查下载的数据库是否完整
for i in {00..55};do md5sum -c nr.${i}.tar.gz.md5;done
2. blast +自带脚本进行下载
版本:version 2.12.0+
下载对象:preformatted-database
perl ~/biosoft/blast/ncbi-blast-2.12.0+/bin/update_blastdb.pl --blastdb_version 5 --decompress nr --source gcp --passive
#使用参数说明:
--blastdb_version # Specify which BLAST database version to download (default: 5).
# Supported values: 4, 5
--decompress # Downloads, decompresses the archives in the current working directory, and deletes the downloaded archive to save disk space, while preserving the archive checksum files (default: false).
--source # to download BLAST databases from (default: auto-detect closest location). Supported values: ncbi, aws, or gcp.
--passive # Use passive FTP, useful when behind a firewall or working in the cloud (default: true).
# To disable passive FTP, configure this option as follows: --passive no
3.wget下载
下载对象:preformatted-database
#网址:https://ftp.ncbi.nlm.nih.gov/blast/db/
#自行编写shell脚本进行下载
for i in {00..55}
do
echo "wget -c https://ftp.ncbi.nlm.nih.gov/blast/db/v5/nr.${i}.tar.gz ./" >> preformatted.wget.nr.sh
echo "wget -c https://ftp.ncbi.nlm.nih.gov/blast/db/v5/nr.${i}.tar.gz.md5 ./" >> preformatted.wget.nr.sh
done
ParaFly -c preformatted.wget.nr.sh -CPU 10 2>err &
(2)提取对应物种的taxid:以植物为例
需要使用到的软件安装:
# taxonkit
conda install -c bioconda taxonkit
# csvtk
conda install csvtk
NCBI使用taxid对物种进行划分,所组成的数据库NCBI Taxonomy。
1、查询对应物种类群的taxid
A. NCBI官网手动查询方式
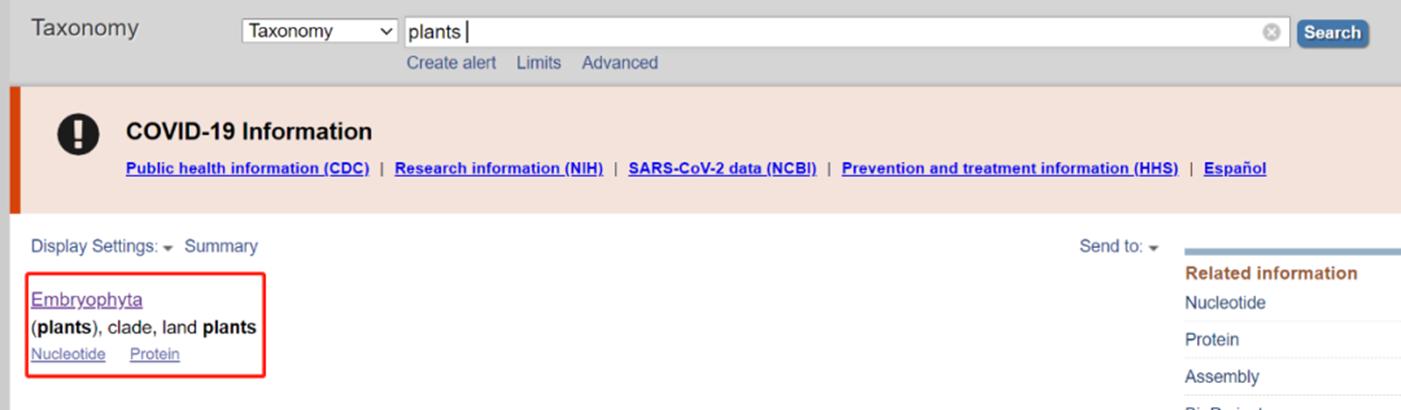
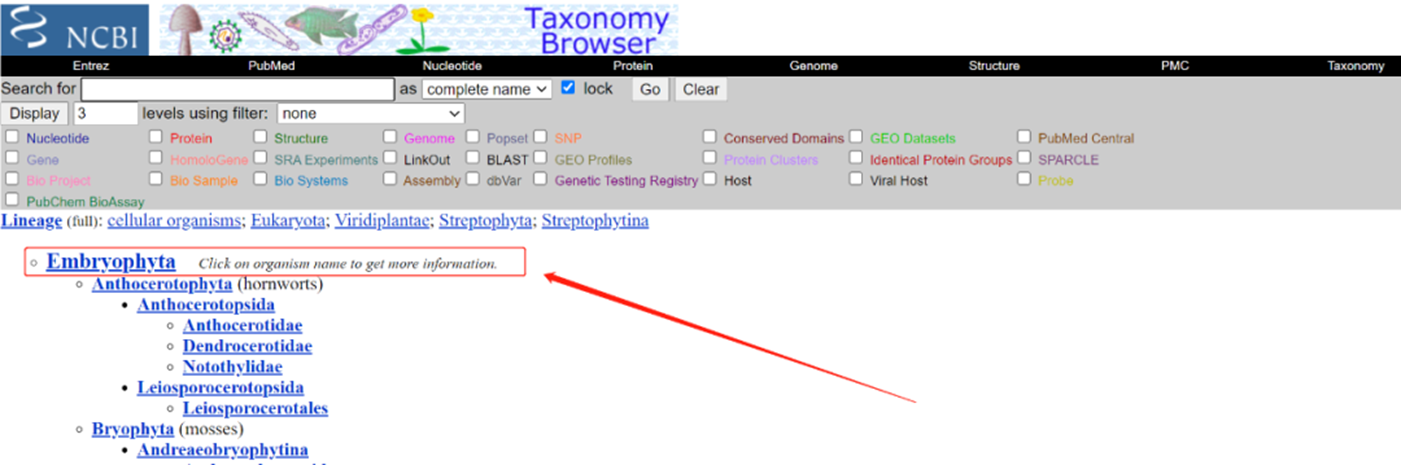
1. 进入NCBI官网,数据库选择“Taxonomy”

1. 输入“Plants”,得到对应查询结果(此处特指为land plants,即陆生植物,绿色植物界包括的范围更大)
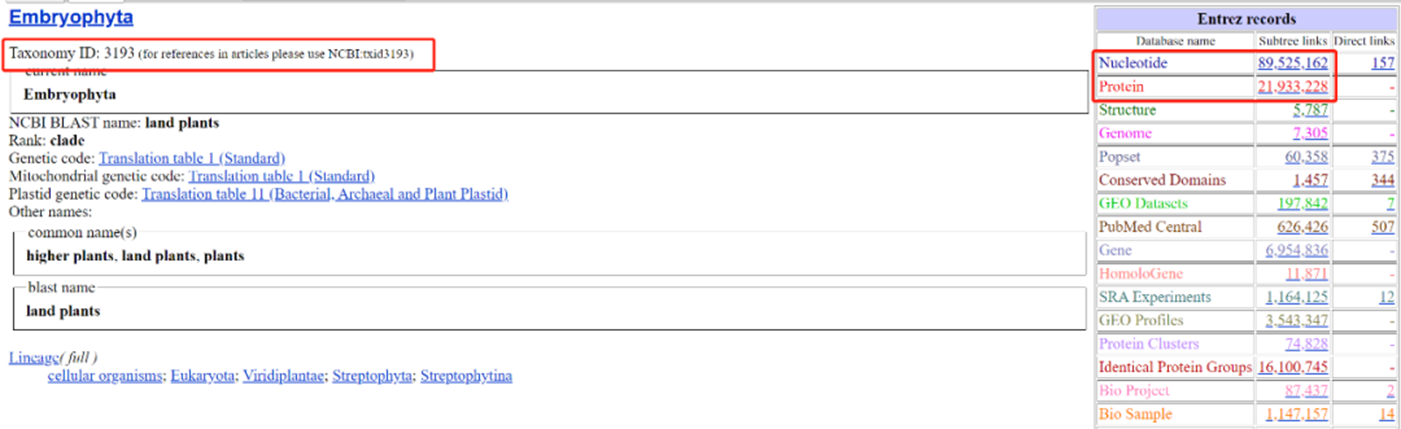
B. taxdump.tar.gz查询
grep -P "\|\s+[pP]lant\w*\s*\|" ~/.taxonkit/names.dmp
C. taxonkit查询
echo Embryophyta | taxonkit name2taxid --show-rank
2、获取对应物种类群所包含的序列(accessionID获取)
1.手动从NCBI官网下载
(1)数据库选择“Protein”。查询格式为txidXXXXX[Organism]或txidXXXXX[ORGN]。
【标注】添加“[]”代表对应taxid所属整体。
(2)查询结果如下:
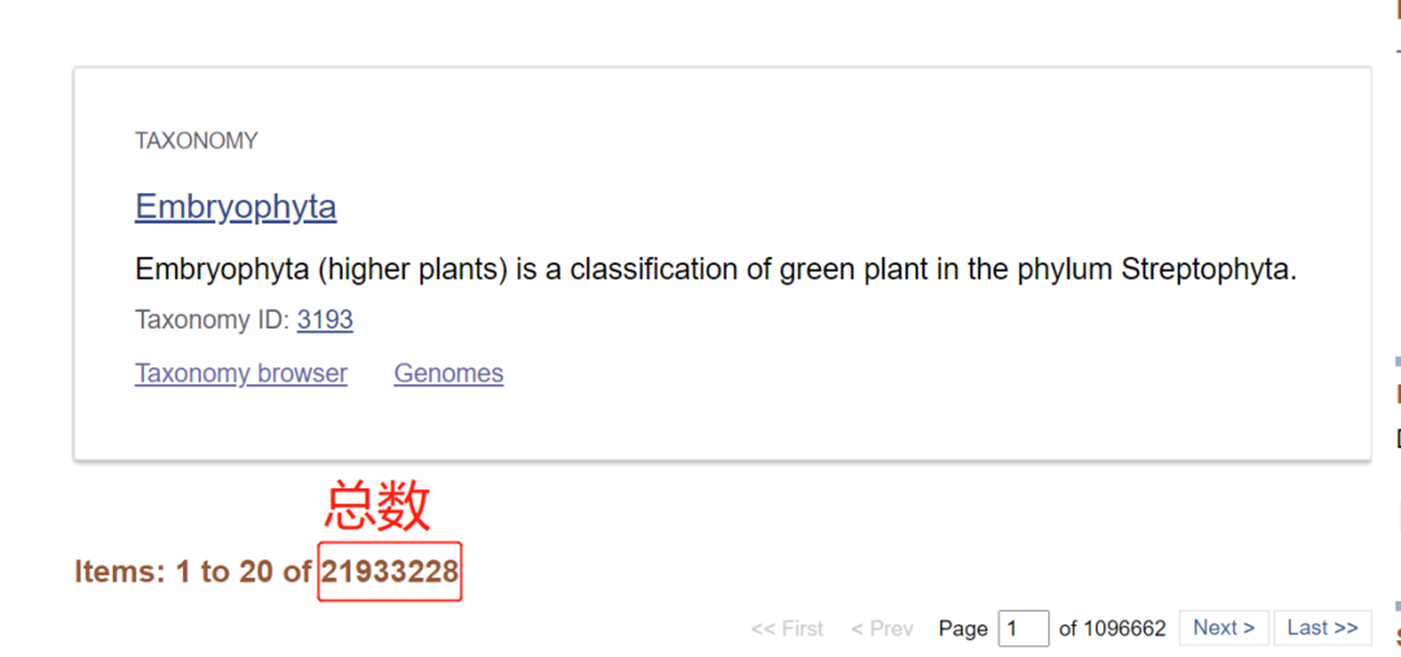
2.使用taxonkit获取对应物种accession ID
需要下载NCBI分类数据&taxid-accession ID转换文件
wget -c https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
wget -c https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz.md5
md5sum -c taxdump.tar.gz.md5 #检查下载数据库是否完整
mkdir ~/.taxonkit
tar zxf taxdump.tar.gz -C ~/.taxonkit
wget -c https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/prot.accession2taxid.gz
wget -c https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/prot.accession2taxid.gz.md5
md5sum -c prot.accession2taxid.gz.md5
#获取对应类群的taxids
taxonkit list --ids 3193 --indent "" > plants.taxid.txt
#获取对应类群的accession ID
pigz -dc prot.accession2taxid.gz | csvtk grep -t -f taxid -P plants.taxid.txt | csvtk cut -t -f accession.version,taxid | sed 1d > plant.acc2taxid.txt && cut -f 1 plant.acc2taxid.txt > plant.acc.txt &
3.使用blast相关脚本获取accession ID
若要使用blast自带脚本get_species_taxids.sh,获取对应物种accession ID,需要提前安装程序EDirect command-lineutility,install manual链接为:https://www.ncbi.nlm.nih.gov/books/NBK179288/
get_species_taxids.sh -t 3193 > plants.blast.taxids.txt
#得到上述taxids list之后,结合csvtk即可得到accession ID
3、对应物种类群nr子库构建
【标注】此处构建子库以preformatted nr database为例
#解压下载好的preformatted nr database
for i in {00..55}
do
tar -zxvf nr.${i}.tar.gz -C ../nr_preformatted_20211122_decompress
done
1.blastdb_aliastool构建blast子库
blastdb_aliastool -seqidlist sequence.seq -db nr -out nr_plant -title nr_plant
# sequence.seq为对应类群的accession list
结果生成一个index文件:nr_plant.pal。
2.提取对应类群序列,自行进行构建本地nr数据库子库
方法1:
blastdbcmd -db nr -entry_batch sequence.seq -out - | pigz -c > blastdbcmd.nr.3193.fa.gz
方法2:
#提取preformatted nr数据库中的所有序列
blastdbcmd -db nr -dbtype prot -entry all -outfmt "%f" -out - | pigz -c > nr.fa.gz
time cat <(echo) <(pigz -dc nr.fa.gz) \
| perl -e 'BEGIN{ $/ = "\n>"; <>; } while(<>){s/>$//; $i = index $_, "\n"; $h = substr $_, 0, $i; $s = substr $_, $i+1; if ($h !~ />/) { print ">$_"; next; }; $h = ">$h"; while($h =~ />([^ ]+ .+?) ?(?=>|$)/g){ $h1 = $1; $h1 =~ s/^\W+//; print ">$h1\n$s";} } ' \
| seqkit grep -f 3193.acc.txt -o nr.3193.fa.gz
【有趣的现象】
在提取完成过后,发现blastdbcmd.nr.3193.fa.gz和nr.3193.fa.gz的文件大小不一样(前13G,后4G)。查看序列数量后,发现2个结果文件所包含的序列数是一样的,造成上述结果的原因是官方自带软件提取的结果,序列表头冗余信息过多。
seqkit stats blastdbcmd.nr.3193.fa.gz
file format type num_seqs sum_len min_len avg_len max_len
blastdbcmd.nr.3193.fa.gz FASTA Protein 19,326,820 7,670,152,256 6 396.9 23,040
seqkit stats nr.3193.fa.gz
file format type num_seqs sum_len min_len avg_len max_len
nr.3193.fa.gz FASTA Protein 19,326,820 7,670,152,256 6 396.9 23,040
若下载的为nr.gz(fasta格式),即非提前构建好的nr数据库,需要在本地使用makeblastdb或diamond makedb进行本地数据库构建
参考资料
[1]http://www.chenlianfu.com/?p=2691
[2]https://bioinf.shenwei.me/taxonkit/tutorial/
[2]https://www.burning.net.cn/article/article-59
本文转载自《生信菜鸟团》。如有侵权,请联系删除。

