本文由医信融合团队成员“陈浩然”撰写,已同步至微信公众号“医信融合创新沙龙”与“研究生学生信”,更多精彩内容欢迎关注!


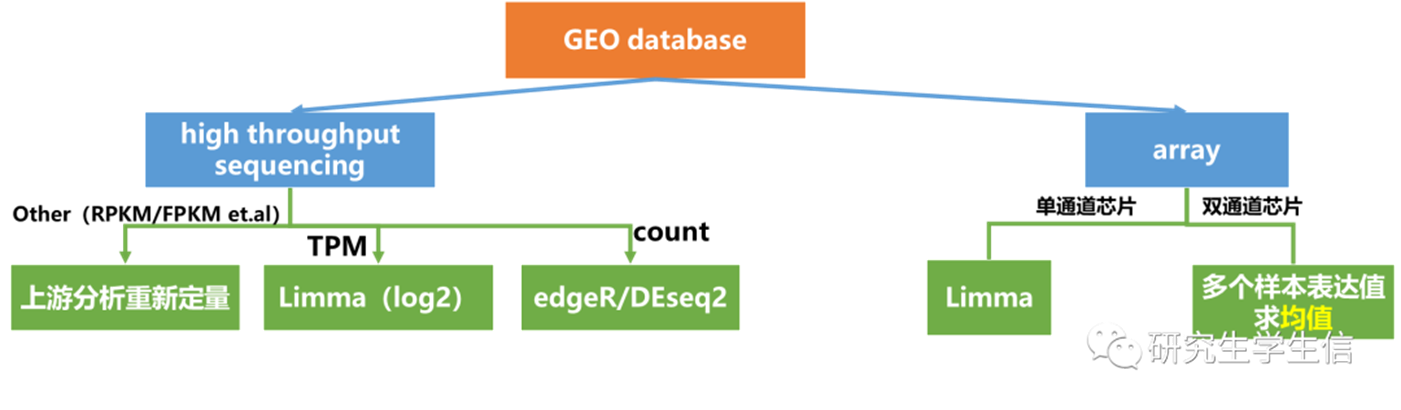
截止2022年4月6日,GEO数据库共包含61,987个高通量测序数据以及65,021个芯片数据,为生信分析、临床研究以及实验设计提供了强有力的数据支撑,今天,主要为大家带来的是GEO数据库的差异分析操作。

一、分析思路选择
二、FAQ
2.1如何判断双通道芯片(例:GSE111471)
1. 双通道芯片的sample title一般为AvsB
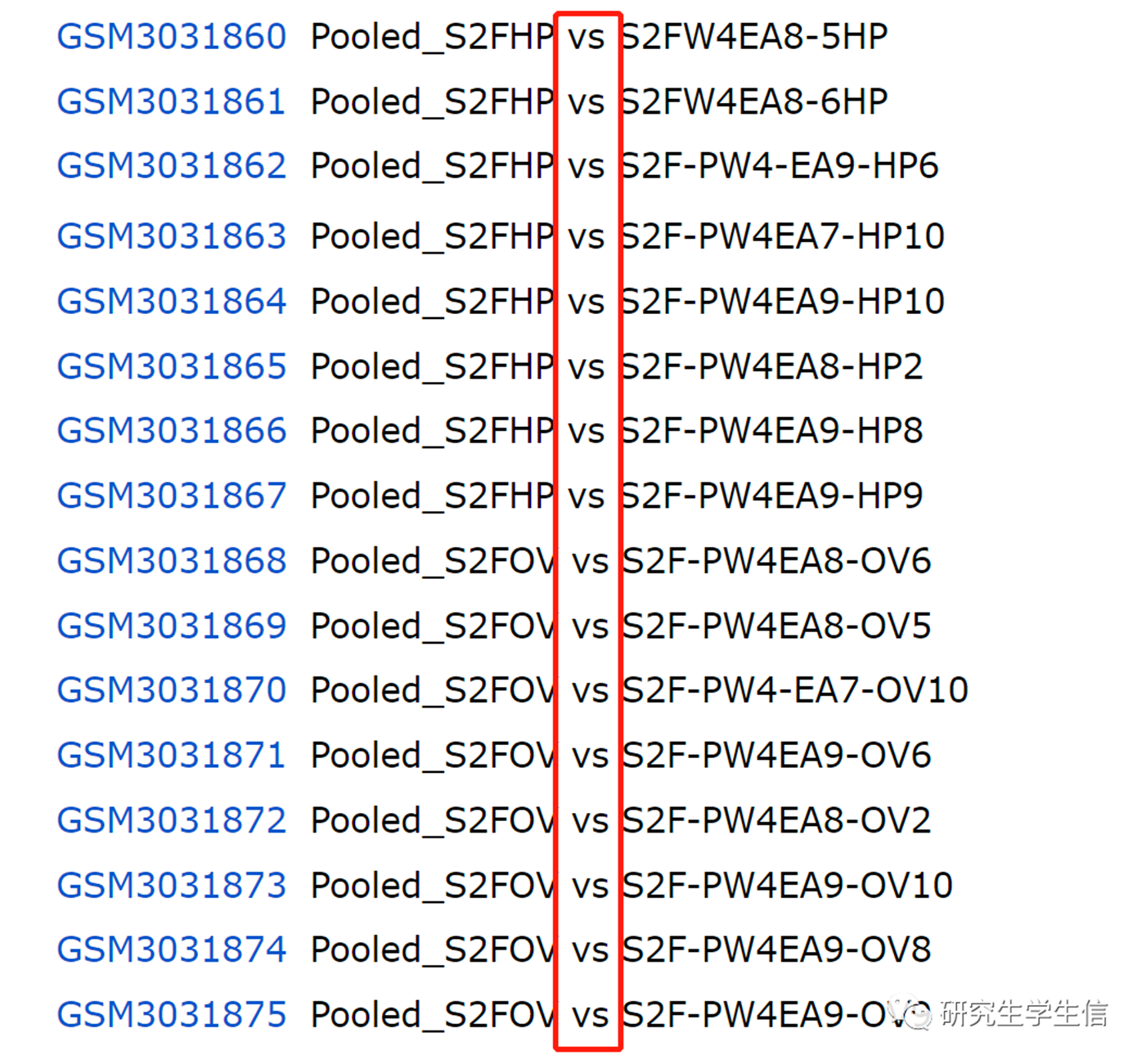
1. 双通道芯片的GSM页面会有两个channel(关于什么是GSM以及GSM在哪,可以查看四文搞定GEO数据库转录组差异分析之简介)

2.2如何判断单通道芯片(例:GSE182065)
不是双通道芯片,就是单通道芯片


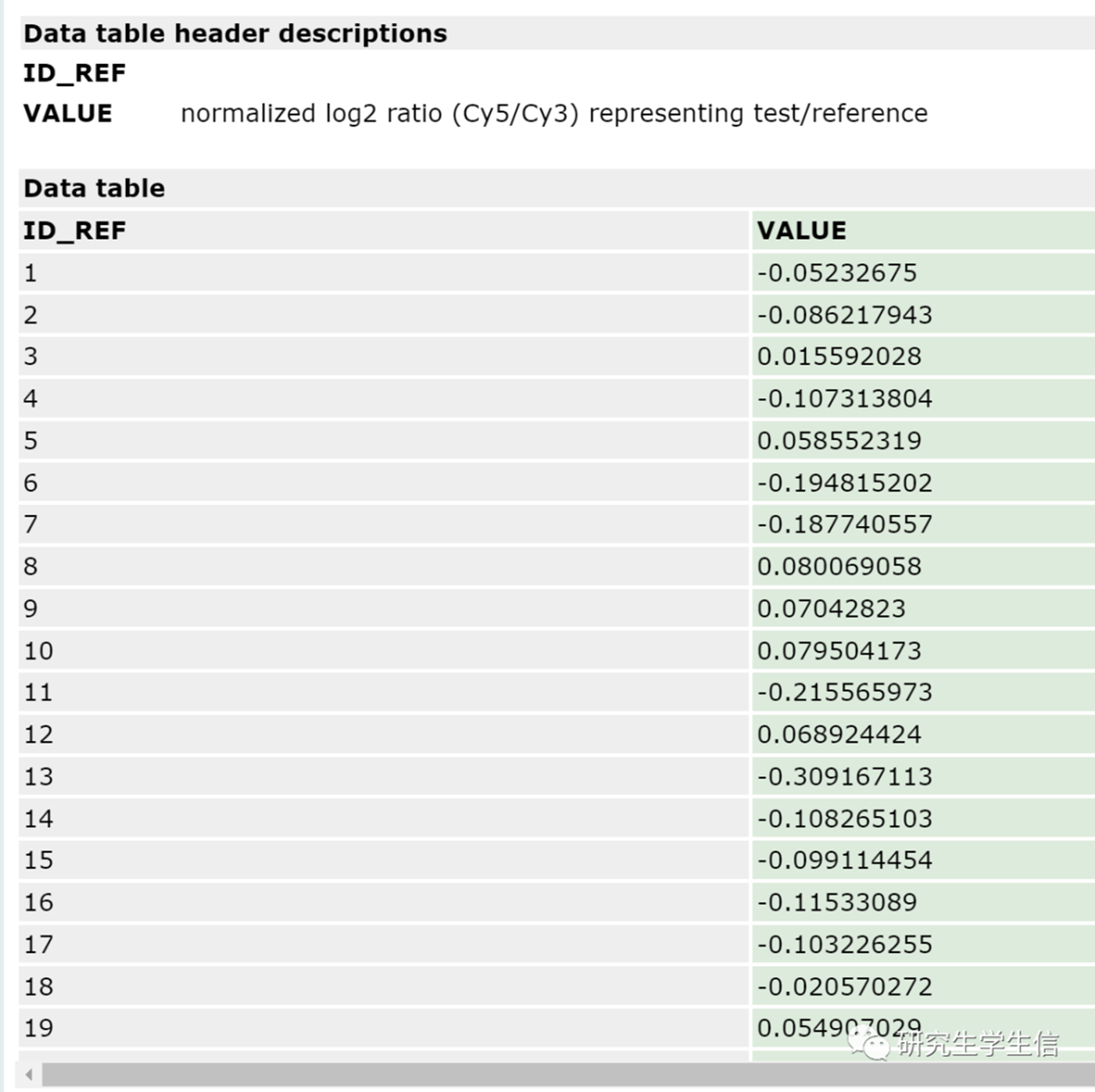
2.3为什么双通道芯片表达量求平均就可以了
如下图所示,双通道芯片原理为两个样本同时测(单通道为一个样一个表达量),其表达量本就是Cy5/Cy3,也就是实验组与对照组的比值(取log2),因此,一般将重复样本之间测量的value求平均后即可作为差异分析的结果。

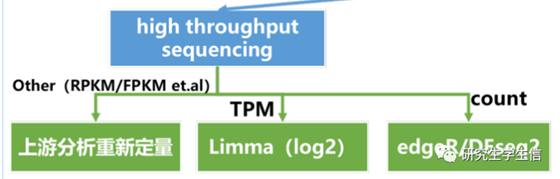
2.4为什么!为什么有了TPM/FPKM/RPKM值不能用limma,还要去做上游分析得到count值,不嫌费劲吗?

友情提示:本问题是给有一定基础的同学看的,现在不理解也可以跳过,回来看的时候记得点赞。

肯定有同学想用limma尝试分析RNA-seq数据了,但是我们不妨再多思考一下

第一种解法:
limma帮助文档**、DEseq2帮助文档、edgeR帮助文档(移动到阅读原文可观看,下不重复)**
学习原始文档,你只需要把这三个帮助文档中几百页的英文单词一个一个翻译出来,把公式弄懂,你就肯定明白了!是不是特别简单

第二种解法:
已知:
1. RPKM, FPKM and TPM, clearly explained(先了解基础知识)
2. 如果咱们有count矩阵,那么最优解就是edgeR和DEseq2
3. 如果是TPM/FPKM/RPKM值等,目前最优解不知道
4. count值可以转为FPKM/TPM/RPKM
求证:
表达矩阵如果为TPM/FPKM/RPKM值如何差异分析?
答1:如果我们有count值,且count可以转为FPKM,有没有一种可能,咱们比较一下count和FPKM差异分析结果的区别?
那么刚好我就有一个count数据(GSE140844),做出来是什么效果呢?
首先说明一下分析方法:
原始数据的count值用的是edgeR和DEseq2两种算法,
count转为FPKM后,用的是limma算法
阈值:logFC>0,**P<0.05**
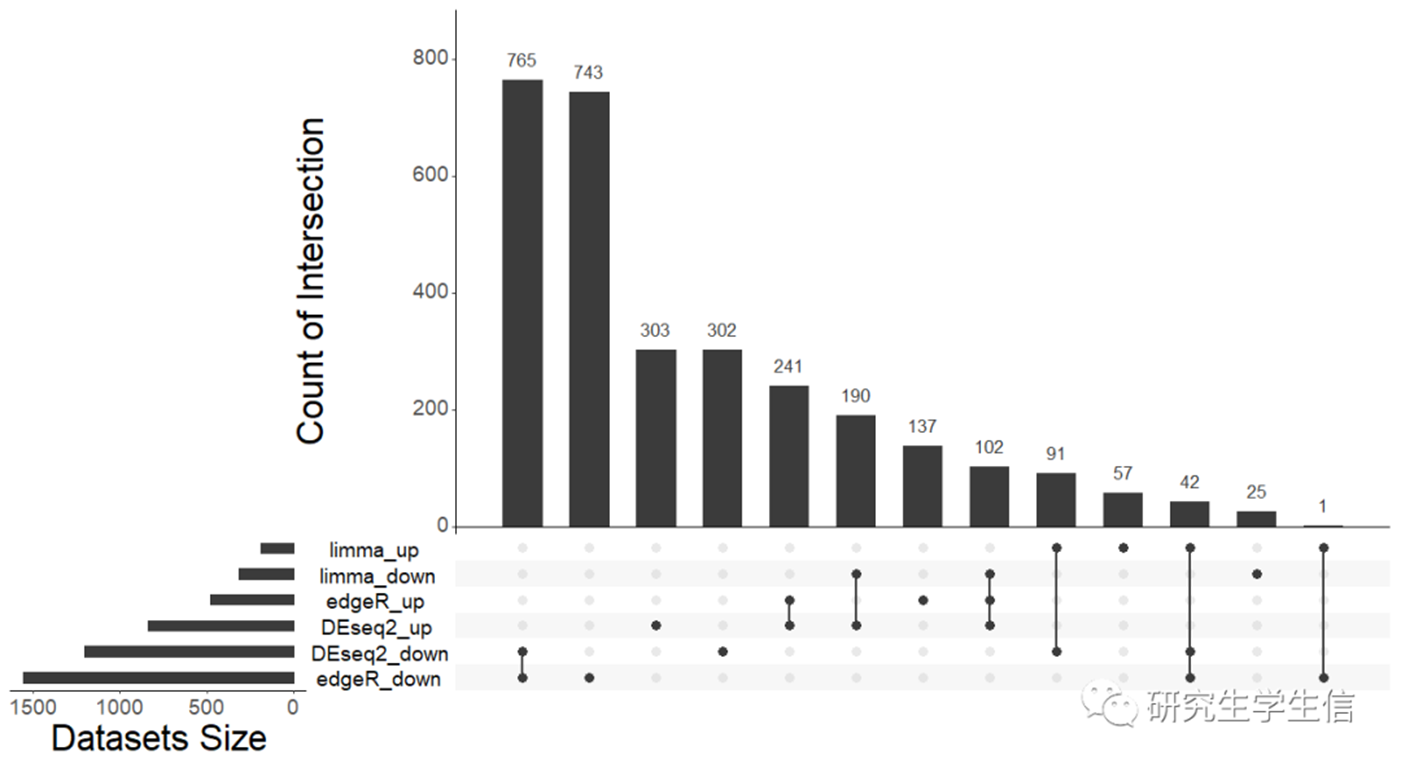
可以看出,FPKM值做limma的结果:
1. 筛选出的差异基因少
2. 和主流算法的交集少的可怜
3. 甚至还有与DEseq2/edgeR调控方向相反的结果出现
答2:如果我们有count值和FPKM值,且count和FPKM均可以转为TPM,有没有一种可能,咱们比较一下TPM和count差异分析结果的区别

那么刚好我就有一个数据GSE162734**(count+FPKM都有)**,做出来是什么效果呢?
首先说明一下分析方法:
原始数据的count值用的是edgeR和DEseq2两种算法,
count/FPKM转为TPM后,用的是limma算法
阈值:logFC>0,**P<0.05**
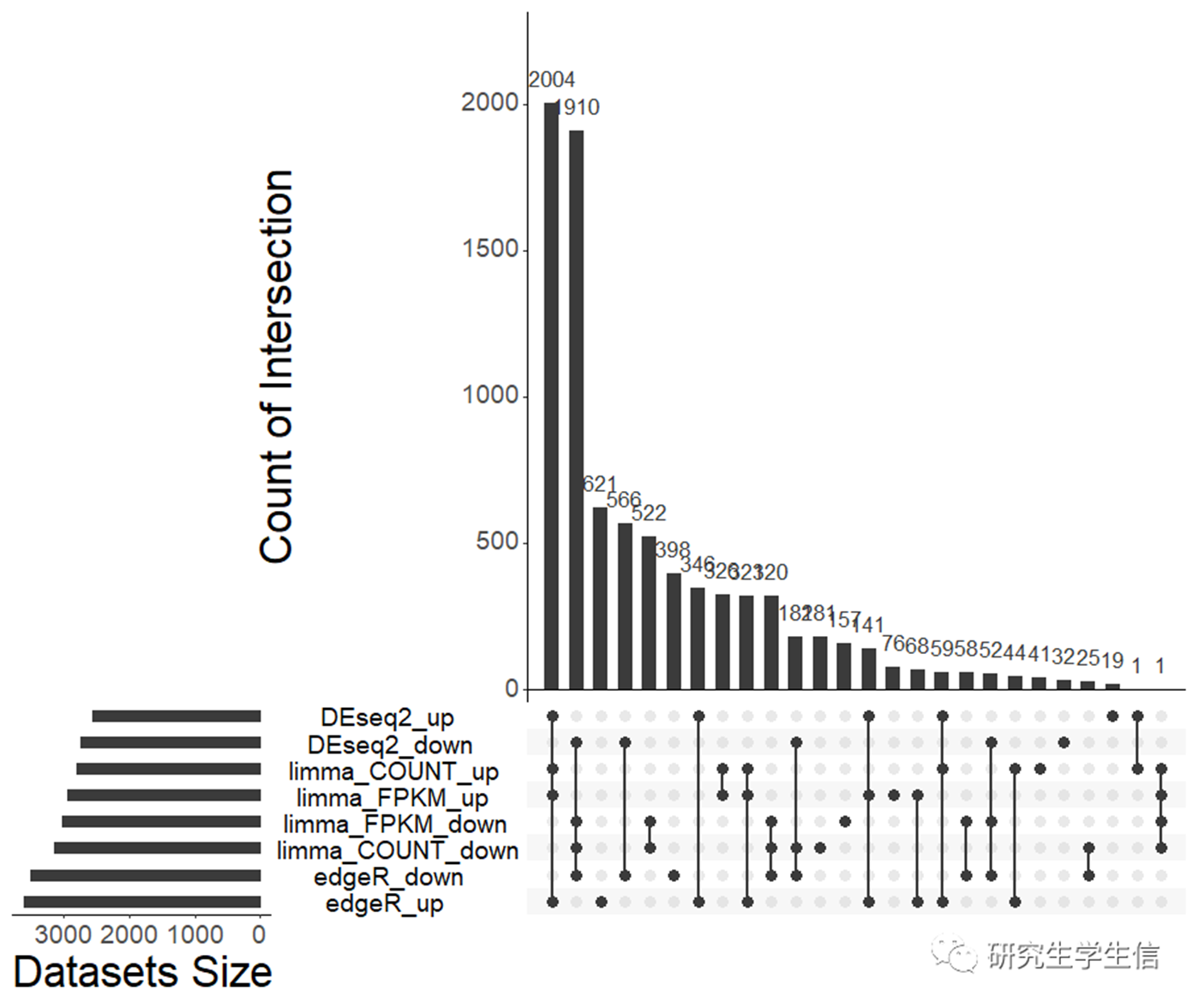
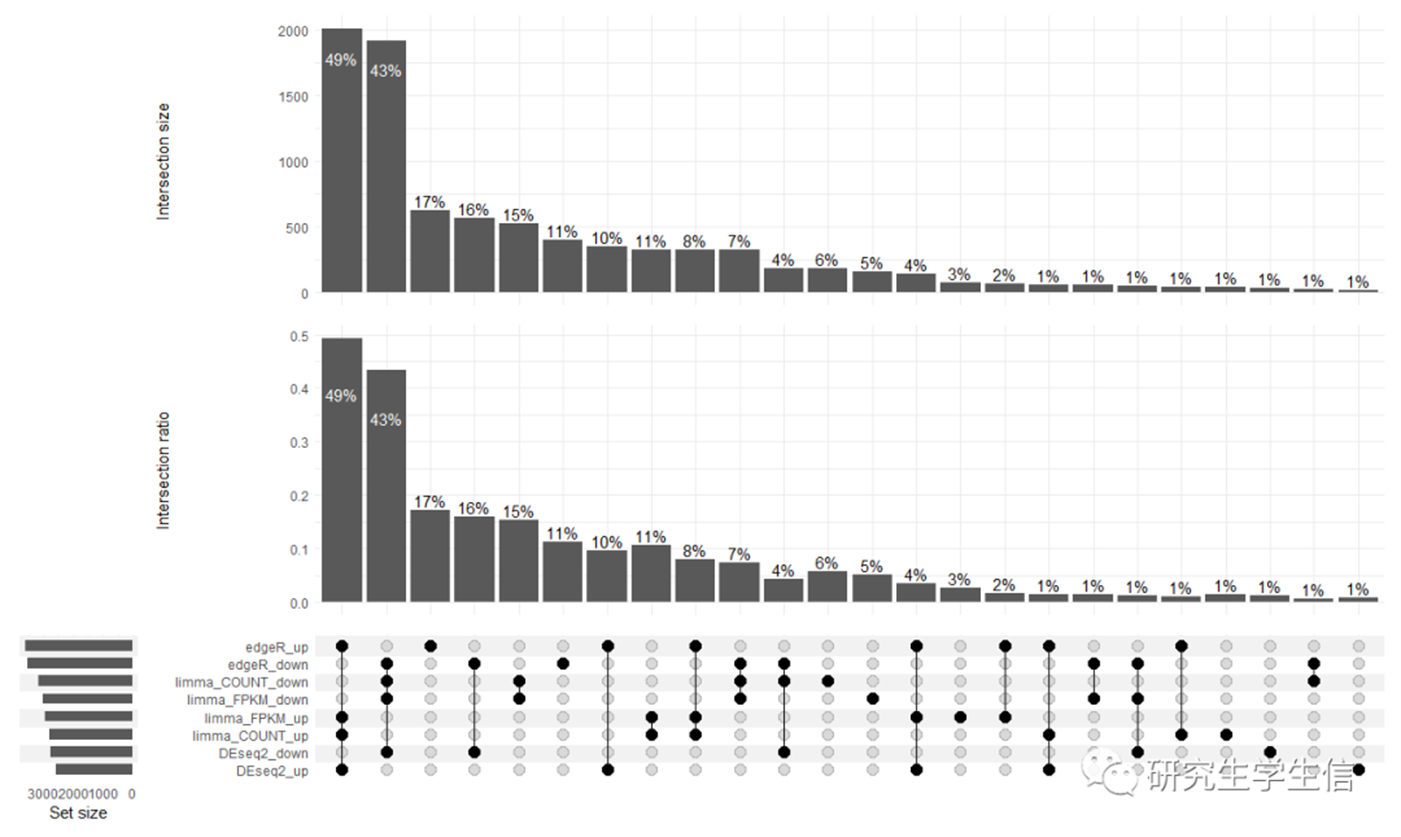
可以看出,TPM值做limma的结果:
1. 筛选出的差异基因与count分析结果类似
2. 交集数量> 90%
3. 交集方向基本相同
总结:
三、GEO转录组数据处理



3.1 array差异分析
3.1.1双通道array差异分析(GSE33812)
需要用到两个文件:
两个文件:GSE的series matrix文件,GPL的annotation或full table文件,两个文件下载位置可参考四文搞定GEO数据库转录组差异分析之简介
一句话介绍我们要干什么:
如下图所示,用GPL4133中ID-gene的对应关系来注释GSE38812的ID列,并且对GSE38812中进行差异分析,得到的结果即为差异结果。
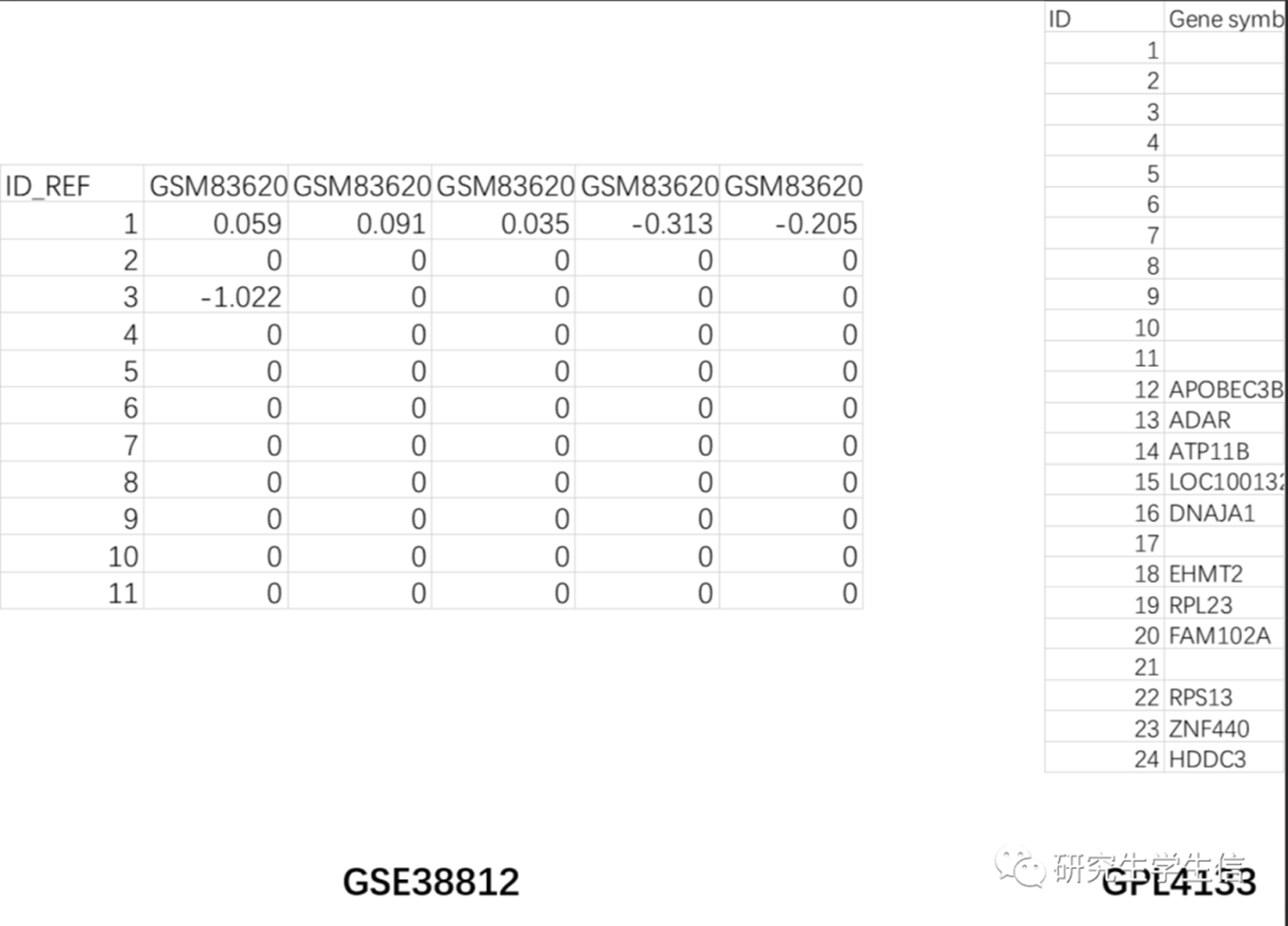
准备的文件
下载GPL4133的annot文件或者full table皆可
将GPL中包含#和!的行都删除,并只保留ID列和Gene symbol列(如上图表格中GPL4133所示),保存到ann.txt中,GSE文件不用动
#现在已默认已下载了series matrix和ann.txt文件到D盘的tem文件夹中
setwd("D:\\tem") #双斜杠或反斜杠(/)分割
ann <- read.table("ann.txt",comment.char = "",header = T,
sep = "\t",quote = "",fill = T)
probeMatrix <- read.table("GSE33812_series_matrix.txt.gz",sep = "\t",
header = T,comment.char = "!")
geneMatrix <- merge(ann,probeMatrix,by.x = "ID",by.y="ID_REF") %>%
.[,c(2:length(.))] %>%
.[.[,1]!="",]
geneMatrix <- aggregate(.~`Gene.symbol`, data=geneMatrix, mean)
rownames(geneMatrix) <- geneMatrix[,1]
geneMatrix <- geneMatrix[,2:ncol(geneMatrix)]
gene_mean <- apply(geneMatrix,1,mean)
output <- cbind(rownames(geneMatrix),gene_mean)
colnames(output) <- c("genes","logFC")
write.table(output,"GSE33812_diff.txt",sep = "\t",row.names = F,col.names = T,quote = F)
3.1.2单通道array差异分析
准备文件:
注:GPL文件中仅需保留ID列和Gene Symbol列,当然如果能完整理解以下代码的,也不用这样严格,之后再删也行


# if (!require("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
# BiocManager::install("limma")
# BiocManager::install("impute")
# install.packages("ggthemes")
# install.packages("dplyr")
#包仅需装一次
rm(list = ls())
library(limma) #载入limma包
library(impute)
library(dplyr)
library(ggthemes)
logFoldChange=1 #设置logFC
P.adjust = T #是否矫正P值
log = T #表达量是否取log值
adjustP = 0.05 #设置P/P.adjust的阈值
test_num <- 16 #实验组样本数量
control_num <- 16 #对照组样本数量
setwd("C:\\Users\\13415\\Desktop\\input") #设置工作目录
ann <- read.table("GPL570-55999.txt", #设置GPL文件
sep = "\t",
header = T,
check.names = F)
probeMatrix <- read.table("GSE5081_series_matrix.txt.gz", #设置series文件,不用解压都可
sep = "\t",
header = T,
check.names = F,
comment.char = "!")
geneMatrix <- merge(ann,probeMatrix,
by.x = "ID", #by.x为注释文件中ID的名称
by.y="ID_REF") %>% #by.y为series文件中ID的名称
.[,c(2:length(.))] %>%
.[.[,1]!="",]
rt=as.matrix(geneMatrix)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
exp=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
exp <- exp[apply(exp,1,mean)>0,]
#impute missing expression data
mat=impute.knn(exp)
rt=mat$data
rt=avereps(rt)
#normalize
pdf(file="rawBox.pdf")
boxplot(rt,col = "blue",main = "Before normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
dev.off()
rt=normalizeBetweenArrays(as.matrix(rt))
pdf(file="normalBox.pdf")
boxplot(rt,col = "red",main = "Normalization",
xlab = "Sample list",
ylab = "Expression value",xaxt = "n",outline = F)
dev.off()
qx <- as.numeric(quantile(rt, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0)
if (LogC) { rt[which(rt <= 0)] <- NaN
rt <- log2(rt) }
#differential
class <- c(rep("dis",test_num),rep("con",control_num))
design <- model.matrix(~0+factor(class))
colnames(design) <- c("con","dis")
fit <- lmFit(rt,design)
cont.matrix<-makeContrasts(dis-con,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='BH',number=200000)
write.table(allDiff,file="limmaTab.xls",sep="\t",quote=F)
#write table
diffSig <- allDiff[with(allDiff, (abs(logFC)>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffSig,file="diff.xls",sep="\t",quote=F)
diffUp <- allDiff[with(allDiff, (logFC>logFoldChange & adj.P.Val < adjustP )), ]
write.table(diffUp,file="up.xls",sep="\t",quote=F)
diffDown <- allDiff[with(allDiff, (logFC<(-logFoldChange) & adj.P.Val < adjustP )), ]
write.table(diffDown,file="down.xls",sep="\t",quote=F)
#write expression level of diff gene
hmExp=rt[rownames(diffSig),]
diffExp=rbind(id=colnames(hmExp),hmExp)
write.table(diffExp,file="diffExp.txt",sep="\t",quote=F,col.names=F)
#volcano
if (P.adjust) {
allDiff$threshold[allDiff$P.Value < 0.05 & allDiff$logFC > logFoldChange ] = "up"
allDiff$threshold[allDiff$P.Value < 0.05 & allDiff$logFC < -logFoldChange ] = "down"
allDiff$threshold[allDiff$P.Value > 0.05 & (allDiff$logFC>= -logFoldChange | allDiff$logFC <= logFoldChange)] = "non"
p_result<- ggplot(allDiff,aes(x=logFC,y=-log10(P.Value),colour=threshold))+xlab("log2 Fold Change")+ylab("-log10adj.P-Value")+
geom_point(size=4,alpha=0.6)+
scale_color_manual(values =c("#0072B5","grey","#BC3C28")) +
geom_hline(aes(yintercept=-log10(0.05)),colour="grey",size=1.2 ,linetype=2) + #增加水平间隔线
geom_vline(aes(xintercept=0), colour="grey",size=1.2 ,linetype=2)+ #增加垂直间隔线
theme_few()+theme(legend.title = element_blank()) #去掉网格背景和图注标签
}else{
allDiff$threshold[allDiff$adj.P.Val < 0.05 & allDiff$logFC > logFoldChange ] = "up"
allDiff$threshold[allDiff$adj.P.Val < 0.05 & allDiff$logFC < -logFoldChange ] = "down"
allDiff$threshold[allDiff$adj.P.Val > 0.05 & (allDiff$logFC>= -logFoldChange | allDiff$logFC <= logFoldChange)] = "non"
p_result<- ggplot(allDiff,aes(x=logFC,y=-log10(adj.P.Val),colour=threshold))+xlab("log2 Fold Change")+ylab("-log10P-Value")+
geom_point(size=4,alpha=0.6)+
scale_color_manual(values =c("#0072B5","grey","#BC3C28")) +
geom_hline(aes(yintercept=-log10(0.05)),colour="grey",size=1.2 ,linetype=2) + #增加水平间隔线
geom_vline(aes(xintercept=0), colour="grey",size=1.2 ,linetype=2)+ #增加垂直间隔线
theme_few()+theme(legend.title = element_blank()) #去掉网格背景和图注标签
}
pdf(file="vol.pdf")
p_result
dev.off()
注意:以上例子假设基因表达谱矩阵除ID_REF列以外,前16列(连续)为实验组,后16列(连续)为对照组
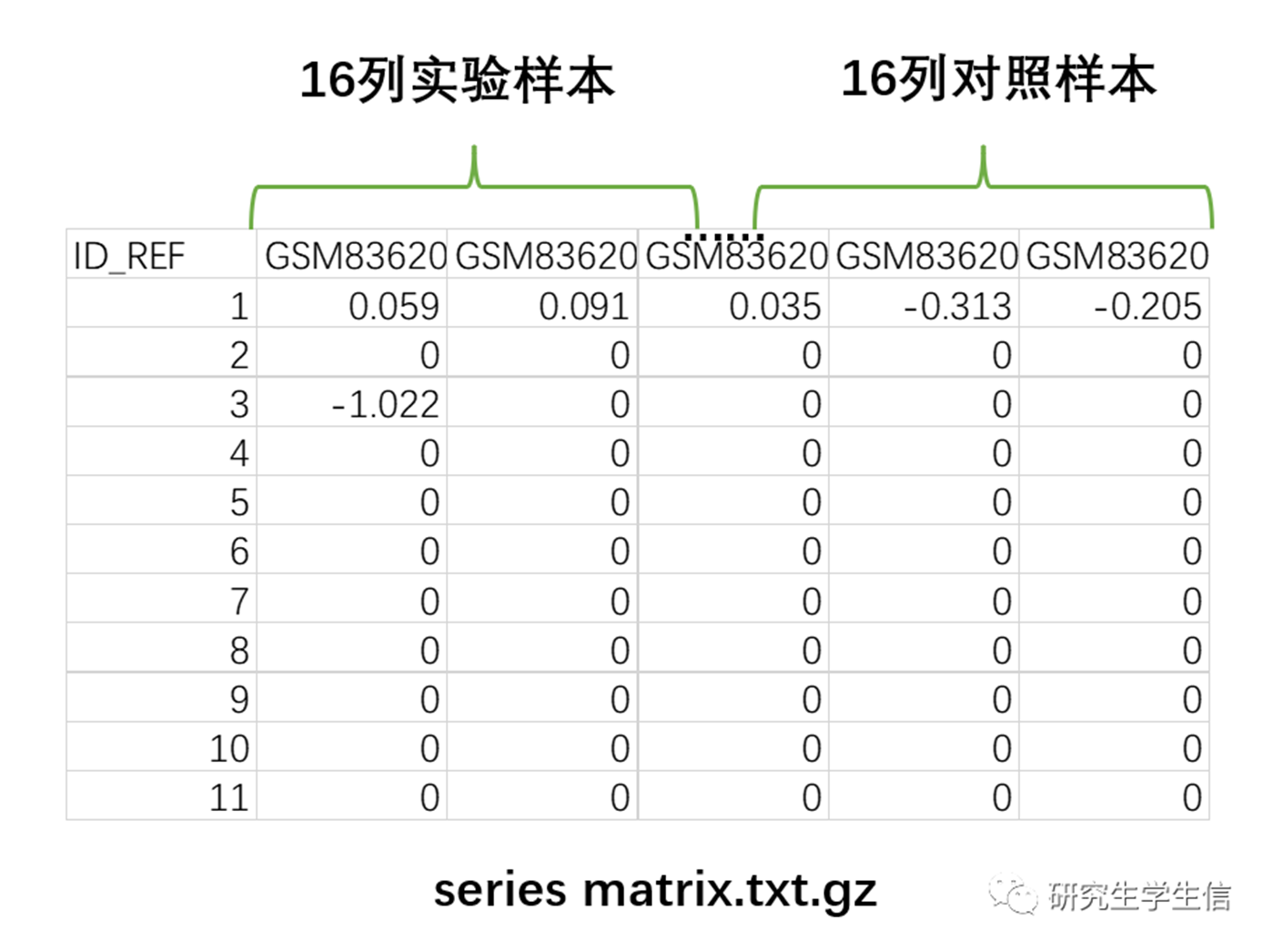
如果你不想解压series matrix.gz的话,可以在R中修改probeMatrix变量的列的顺序(选择性运行)
probeMatrix <- probeMatrix[,c(18:33,2:17)]
#上面例子为,将probeMatrix的18-33列移动到2-17列前面(得考虑第一列为ID_REF)
probeMatrix <- probeMatrix[,c(4,5,6,1,2,3)]
#上面例子为,将probeMatrix的456列移动到123列前面
最终效果图:
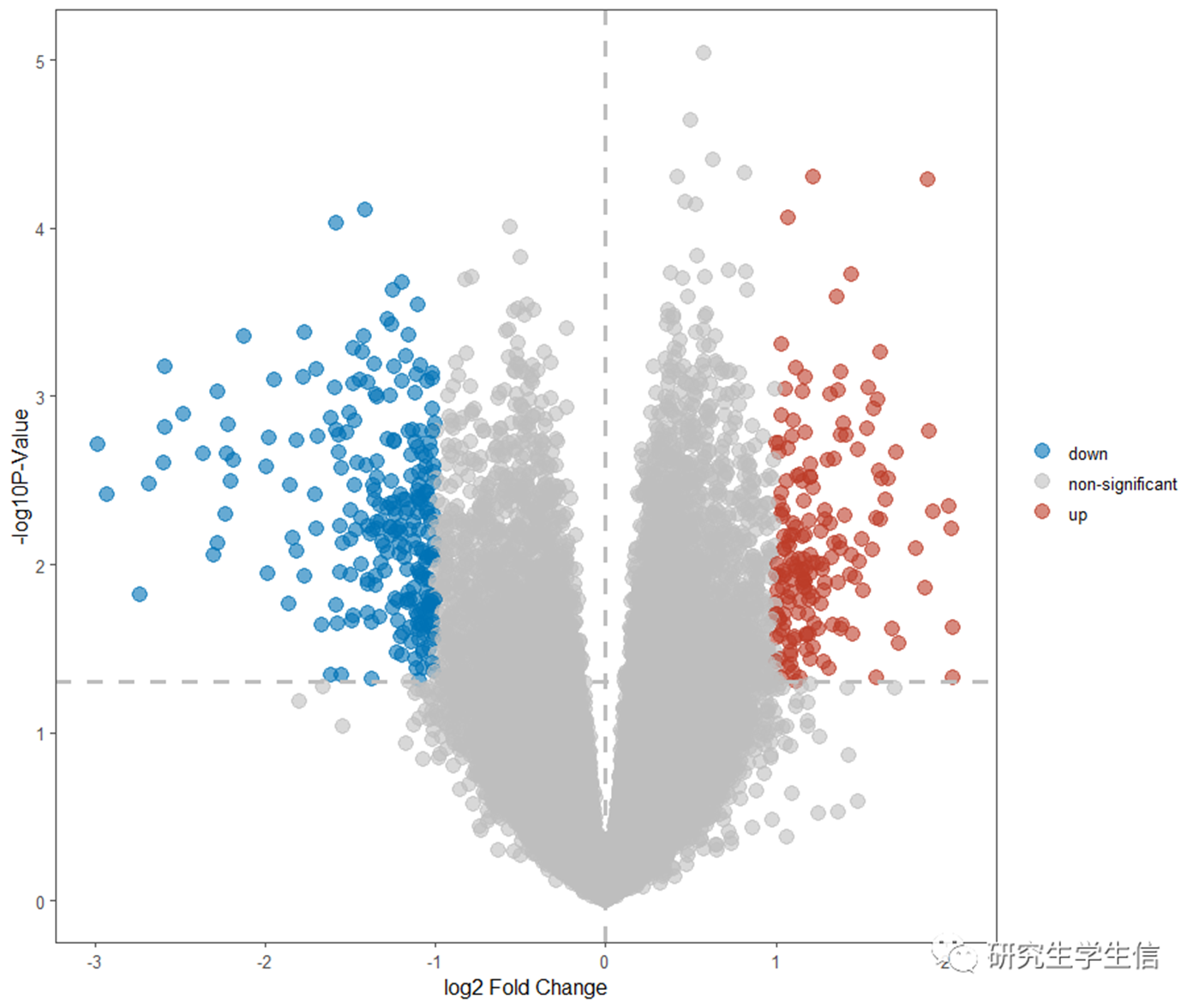
3.1.3 High throughput sequencing
1. 所有的非count、非TPM标准化格式的数据,果断从fasq开始重新比对、定量(上游分析)
2. 本文并未介绍上游分析相关内容,有需要可以点赞、点击在看催更,点赞100+更新buxianggeng
3. TPM的数据使用单通道array差异分析中的流程即可
4. 本小节仅涉及count格式数据的DEseq2算法的分析流程
5. GEO转录组数据分析并不包含ID转换的相关教程,有需要可以点赞、点击在看催更,点赞100+更新buxianggeng
# BiocManager::install("DESeq2")
rm(list = ls())
library(DESeq2)
library(limma)
library(edgeR)
library(ggplot2)
library(dplyr)
library(tinyarray)
logFC_cutoff = 0 #筛选LogFC值
P_adj = F #是否矫正P值
adj_Pvalue = 0.05 # if P_adj = F, the adj_Pvalue just means P_value
ncontrol = 3 #控制组样本数量
ntreat = 3 #实验组样本数量
setwd("E:\\tem") #设置工作路径
rt<-allfile_matrix
library(limma)
rt<-avereps(rt)
rt <- aggregate(.~genes,mean,data=rt)
rownames(rt) <- rt[,1]
rt <- rt[,-1]
exprSet <- round(rt)
group_list <- factor(c(rep("N",ncontrol), rep("T",ntreat)))
colData <- data.frame(row.names=colnames(exprSet), group_list=group_list)
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
res <- results(dds, contrast=c("group_list","T","N"))
DEG=as.data.frame(res)
DEG = na.omit(DEG)
if (P_adj == T) {
down = DEG[(DEG$padj < 0.05)&(DEG$log2FoldChange < -logFC_cutoff),]
up = DEG[(DEG$padj < 0.05)&(DEG$log2FoldChange > logFC_cutoff),]
}else{
down = DEG[(DEG$pvalue < 0.05)&(DEG$log2FoldChange < -logFC_cutoff),]
up = DEG[(DEG$pvalue < 0.05)&(DEG$log2FoldChange > logFC_cutoff),]
}
sigdiff <- rbind(up,down)
sigdiff <- cbind(rownames(sigdiff),sigdiff)
colnames(sigdiff)[c(1,3)]<-c("genes","logFC")
DEG2 <- cbind(rownames(DEG),DEG)
colnames(DEG2)[1] <- "genes"
sigdiff <- sigdiff[,c(1,3,2,4,5,6,7)]
DEG2 <- DEG2[,c(1,3,2,4,5,6,7)]
write.table(sigdiff,"diffSig.xls",sep = "\t",row.names = F,col.names = T)
write.table(DEG2,"DEG_all.xls",sep = "\t",row.names = F,col.names = T)
###热图
# cg1 = rownames(sigdiff)
# pdf(file="mrnaHeatmap.pdf",height=12,width=15)
# draw_heatmap(exprSet[cg1,],group_list,n_cutoff = 2)
# dev.off()
###火山图
DEG$log2FoldChange[DEG$log2FoldChange>5]<-5
DEG$pvalue[-log10(DEG$pvalue)>10]<- 10^-10
DEG$padj[-log10(DEG$padj)>10]<-10^-10
if (P_adj == T) {
vol_plot<-draw_volcano(DEG,pkg = 1,logFC_cutoff = logFC_cutoff,adjust = T)+
theme(plot.title = element_text(hjust = 0.5,size = 16),
axis.text=element_text(size=16),
axis.title.x = element_text(size = 16),
legend.title = element_text(size=16),
legend.text = element_text(size=16))+xlab("LogFC")+ylab("-log10(Adjust P-value)")
dev.off()
}else{
vol_plot<-draw_volcano(DEG,pkg = 1,logFC_cutoff = logFC_cutoff,adjust = F)+
theme(plot.title = element_text(hjust = 0.5,size = 16),
axis.text=element_text(size=16),
axis.title.x = element_text(size = 16),
legend.title = element_text(size=16),
legend.text = element_text(size=16))+xlab("LogFC")+ylab("-log10(P-value)")+ggtitle("Volcano")
}
pdf(file="mrnaVol.pdf",height=8,width=8)
plot(vol_plot)
dev.off()
最终效果图:
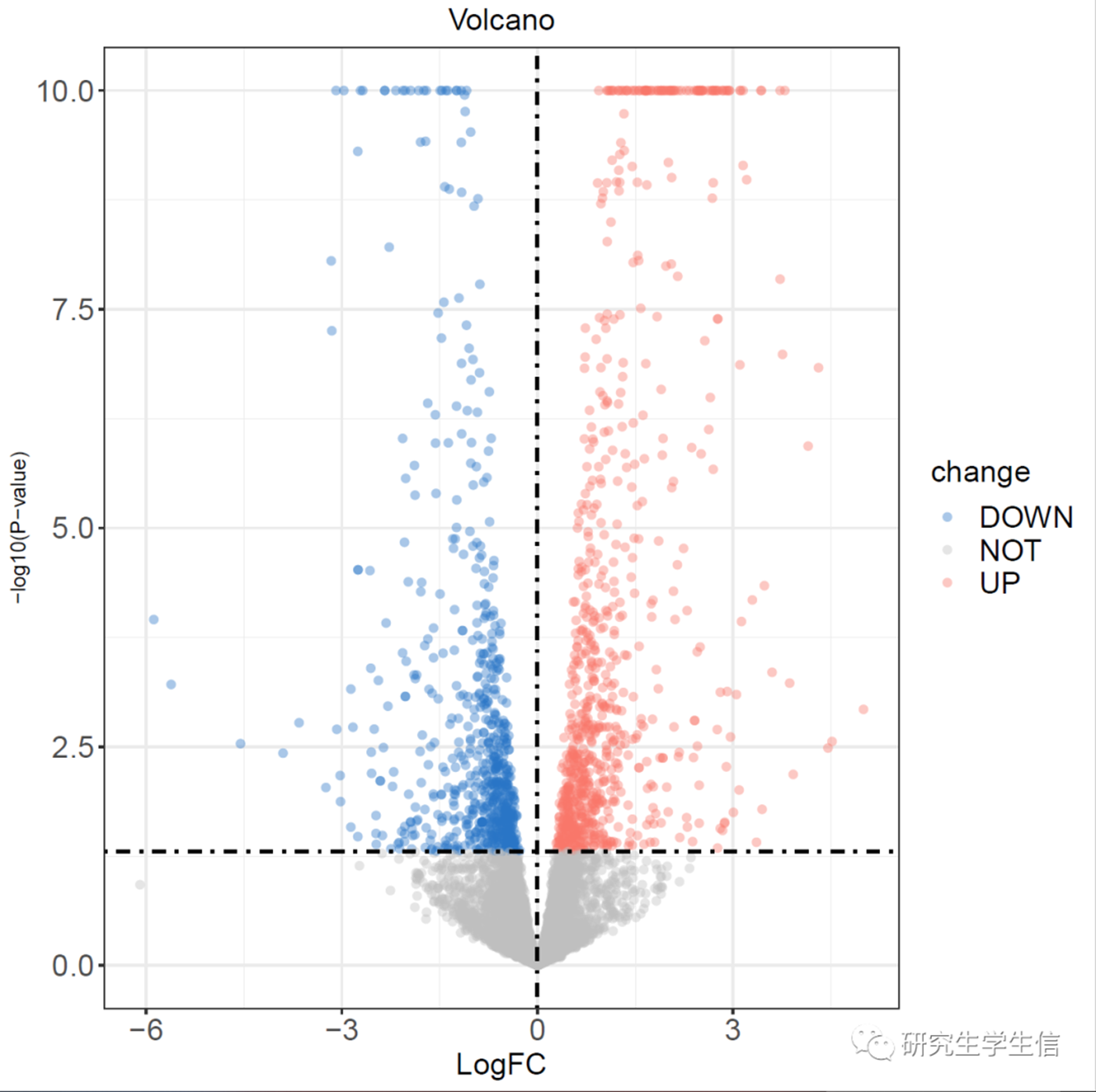
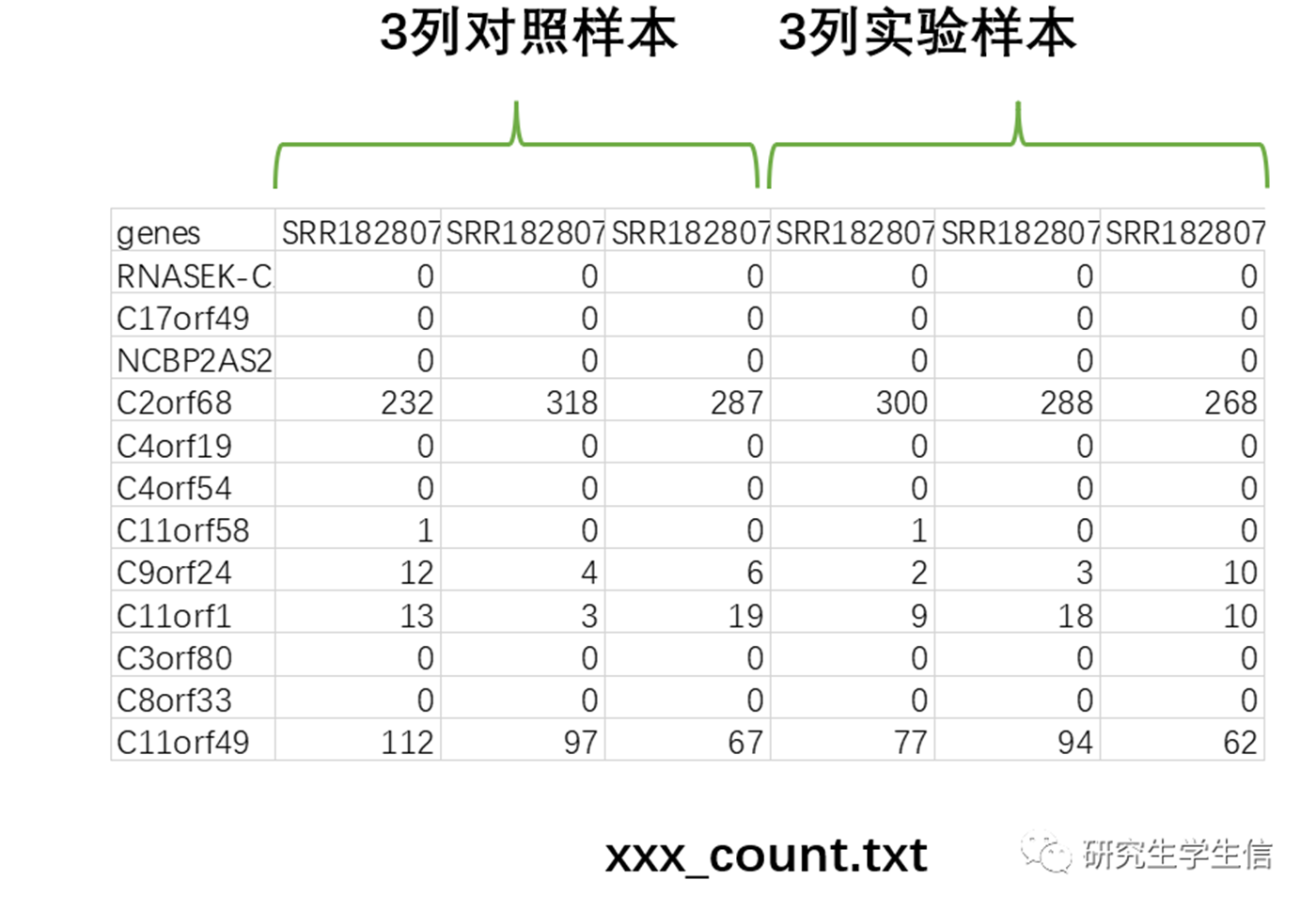
注意:以上例子假设基因表达谱矩阵除genes列以外,前3列(连续)为实验组,后3列(连续)为对照组
四、说在最后
1. 本期待填的坑:
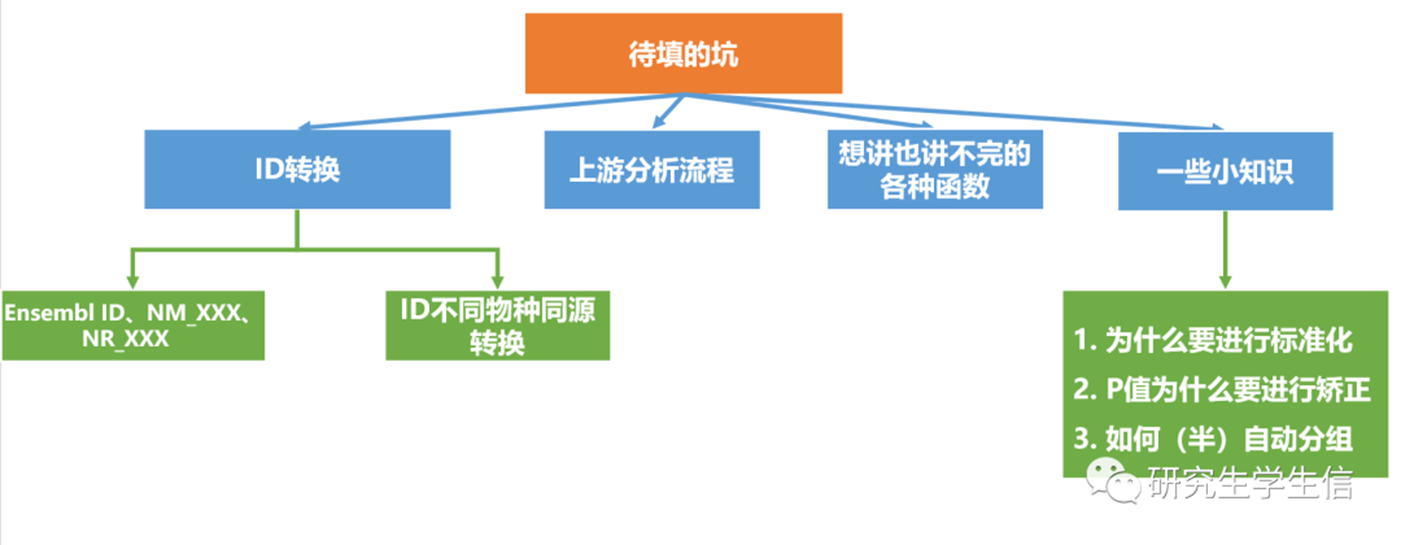
1. 坑都随缘填,如果有志之士想要交流的可后台留言,看见问题会回复
2. 从下期开始总结常见的生物信息分析结果图及其生物学意义
3. 感谢观看到最后

图文:陈浩然
本文编辑:莫状

